Phylogenetics
How to calculate dN, dS, and dN/dS ratio on a set of genes using MEGA?
If you want to get a quick idea about the non-synonymous vs synonymous (dN/dS) substitutions, you can easily use MEGA software [1]. Although HYPHY/Datamonkey provides the best results regarding selection pressure analyses. MEGA also uses HYPHY program [2] to calculate the dN/dS substitutions rate. Here is how you can do it.
You will need a codon fasta file genes, if you have protein sequences, then convert them into nucleotide codon sequences.
i) open MEGA --> Align --> Edit/Build Alignment --> Retrieve sequences from a file. (If you already have an alignment file then skip this step).
ii) Edit --> Select all --> Align by ClustalW/Muscle.
iii) Save the session and export alignment in MEGA format (.meg).
iv) Minimize the alignment window. Go to the main window and click on "Selection" --> "Estimate selection for each codon (Hyphy)".
v) It will prompt to select a .meg file, select it.
vi) It will ask for analysis preferences as shown in Fig. 1. Choose according to your requirements and click "Compute".
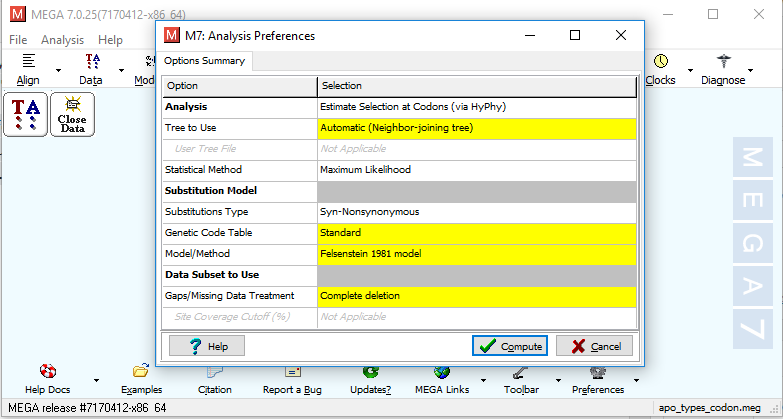
Fig. 1 Analysis preference window for dN/dS substitution calculation using MEGA7.
vii) Later, it will ask for the output format and output directory where you want to save the results. Click "Ok". Your job will be finished after a few minutes depending up on the number and length of sequences.
References
- Kumar, S., Stecher, G., & Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular biology and evolution, 33(7), 1870-1874.
- Pond, S. L. K., & Muse, S. V. (2005). HyPhy: hypothesis testing using phylogenies. In Statistical methods in molecular evolution (pp. 125-181). Springer, New York, NY.


Previously, we have provided an installation tutorial for IQ-TREE on Ubuntu. In this article, we are going to perform model selection for a dataset using the standalone tool of IQ-TREE. (more…)

TREE-PUZZLE is a software to reconstruct phylogenetic trees using the maximum likelihood method [1,2]. It requires sequence data as input and implements a fast search algorithm and quartet puzzling. It can process large datasets easily. In this article, we will install TREE-PUZZLE on Ubuntu. (more…)

MEGAX is a bioinformatics software/tool used for phylogenetic tree construction. In this article, we will construct a maximum likelihood (ML) tree for a number of protein sequences using MEGA7 [1]. (more…)

Covid19 has created a great threat to human health. As you are aware, in this coronavirus outbreak, Bioinformatics Review has created a group, BiR-nCov19 Drug Development Team, to work on finding prevention to this disease. This research group consists of researchers from all over the world. (more…)
A novel coronavirus (CoV), named Severe Acute Respiratory Syndrome-CoV-2 (SARS-CoV-2) or nCoV-2019, has emerged since December 2019 from Wuhan city of Hubei province in China [1]. This virus belongs to the coronavirus family from which previous outbreaks have emerged (SARS and MERS). They have been a great threat to public health causing many deaths including SARS-CoV-2. There is no proper treatment available to cure this coronavirus disease (covid19). Scientists and researchers are trying really hard to develop a drug or a vaccine or a proper way to cure covid19. (more…)

Prottest3 is a software which is used to select a best-fit amino acid replacement model for a set of protein sequences [1]. ProtTest3 finds a best-fit model on the basis of the smallest value of one of three criteria: Akaike Information Criterion (AIC), Corrected Akaike Information Criterion, Bayesian Information Criterion (BIC) score or Decision Theory Criterion (DT) selected by the user. In this article, we will learn how to download and install the command-line version of ProtTest3 on Ubuntu. (more…)

Most of the times, it is a very tedious job to convert file formats in bioinformatics, especially when we are dealing with phylogeny. Most of the available online servers mess your file and the output format is also not supported by the other programs. Additionally, it is quite difficult to perform other customizations on the phylogeny tree. (more…)
Understanding evolution is critical for understanding biology. As the preeminent scientist Theodosius Dobzhansky stated, “Nothing in biology makes sense except in the light of evolution.” Evolution is the only scientific explanation for the diversity of life. It explains the striking similarities among vastly different forms of life, the changes that occur within populations, and the development of new life forms. Excluding evolution from the science curricula or compromising its treatment deprives students of this fundamental and unifying scientific concept to explain the natural world. (more…)

MEGA: Molecular Evolutionary Genetic Analysis
It is important to know the basic molecular relationship between two living organisms as one begins performing comparative studies for knowing the evolutionary aspects and for contributing to knowledge base. Several tools and soft ware have been introduced for meeting the task of such analysis. Each tool has different algorithm and method to perform molecular phylogeny. Examples include; ClustalW, Dendroscope, Hyphy, PAUP and Phylip etc. Among them is the most efficient tool, MEGA, Molecular evolutionary phylogenetic analysis which performs both sequence analysis and phylogenetic analysis in a very sophisticated manner.
(more…)
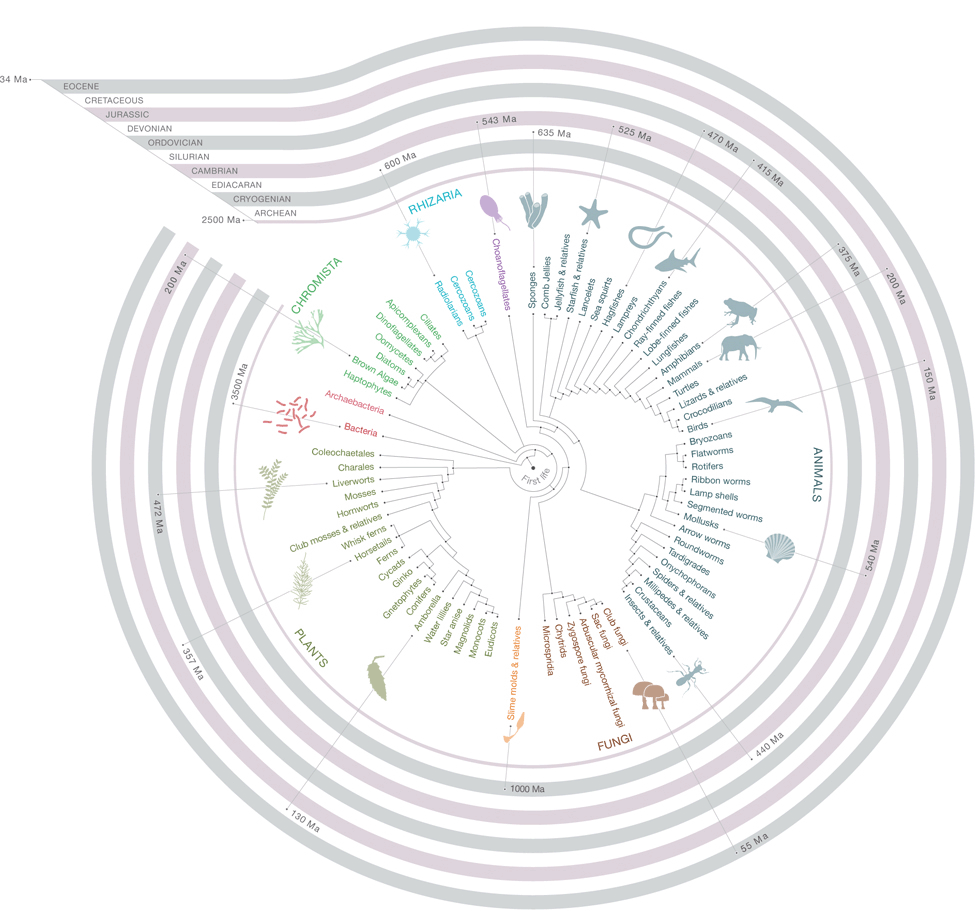
Estimation of present day diversity of organism and understanding their diversity forms new cornerstones of conservation biology, evolutionary biology and ecology. Maintaining the totality of all texa from past to present and classifying into groups reflect how they have changed over the period of times. Necessarily, phylogenetics and evolutionary study of a particular group help out in knowing pattern of occurrence and relationships between two distantly related class or families and their descendents. (more…)

You must be logged in to post a comment Login