MD Simulation
Tutorial: A quick MD simulation using NAMD and VMD

Molecular dynamics (MD) simulation has become an important methodology in research covering systems consisting of millions of atoms. We have provided several articles on MD simulation including the GROMACS [1] installation and performing MD. In this article, we will learn about “QwikMD” a plugin in NAMD [3], and VMD [4].
QwikMD allows users to set up the MD simulation of macromolecules in a few minutes. The steps are explained as follows.
- Open terminal (Ctrl+Alt+T) and type
$ VMD. (If you are using Windows, then open a command prompt by typing ‘cmd’ in the search box and then type>VMD). - Go to
Extensions --> Simulate --> QwikMD. A window will appear as shown in Fig. 1.
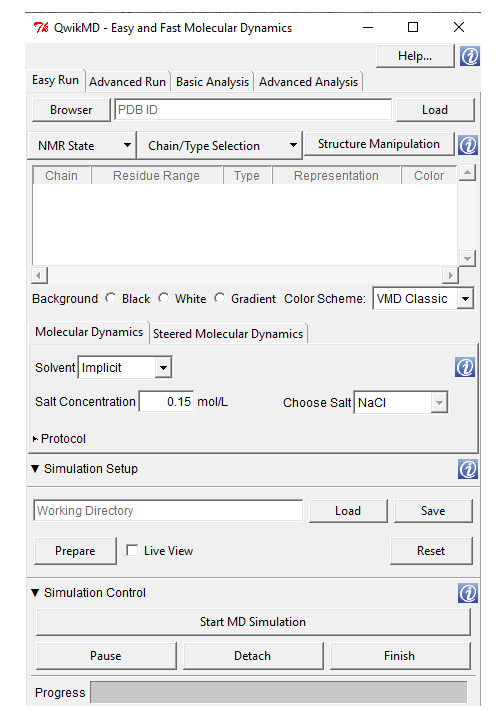
Fig. 1 A screenshot of QwikMD window launched from VMD
3. Now, load your protein structure either by typing or by browsing, then Click 'Load' .
It allows users to either simulate in an explicit solvent by simulating all atoms of the solvent or in an implicit solvent by adding a dielectric constant to the electrostatic calculation. For more information regarding this, click here. We are performing a simple MD and we will be using implicit solvent method.
4. Remove oxygen atoms from water molecules.
Click on 'Chain/Type Selection' and deselect the water molecules 'A and water'.
After that, you can see the oxygen atoms (red) in the VMD display will disappear.
5. Select the ‘Solvent’ and ‘Salt concentration’. Here, it is ‘implicit’ and default salt concentration (0.15 mol/L).
6. Now, move to the section ‘Simulation Set up’.
Check the 'Live View' checkbox for Live simulation and click 'Prepare'.
During this step, autopsf function is called to generate files required for MD simulation including NAMD configuration files. You will have to select a folder to save the input file named ‘.qwikmd‘.
After that, you will notice two folders will be created in the working directory: setup and run. All files created during the set up will be stored in the setup folder and those files that will be required for running MD simulation will be stored in run folder.
7. Move to another option “Protocol”.
Check the 'Equilibration' and 'MD' checkboxes and enter particular 'Temperature' and 'Simulation time'.
Here, the temperature was set to 27 C (300 K) and the simulation time was set to 1ns.
8. Run the simulation
Click 'Start MD Simulation'--> Select the number of CPU cores to be used to perform the simulation.
It might show an error (” Error connecting to localhost on port 3000“) until a connection is established between NAMD and VMD. The live simulation will be continued to run in the background until you abort it.
That’s it. Wait for the simulation to finish.
References
- Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., & Lindahl, E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1, 19-25.
- Ribeiro, J. V., Bernardi, R. C., Rudack, T., Schulten, K., & Tajkhorshid, E. (2018). QwikMD-Gateway for Easy Simulation with VMD and NAMD. Biophysical Journal, 114(3), 673a-674a.
- Phillips, J. C., Zheng, G., Kumar, S., & Kalé, L. V. (2002, November). NAMD: Biomolecular simulation on thousands of processors. In SC’02: Proceedings of the 2002 ACM/IEEE conference on Supercomputing (pp. 36-36). IEEE.
- Humphrey, W., Dalke, A., & Schulten, K. (1996). VMD: visual molecular dynamics. Journal of molecular graphics, 14(1), 33-38.













Bioinformatics Programming
Free_Energy_Landscape-MD: Python package to create Free Energy Landscape using PCA from GROMACS.

In molecular dynamics (MD) simulations, a free energy landscape (FEL) serves as a crucial tool for understanding the behavior of molecules and biomolecules over time. It is difficult to understand and plot a meaningful FEL and then extract the time frames at which the plot shows minima. In this article, we introduce a new Python package (Free_Energy_Landscape-MD) to generate an FEL based on principal component analysis (PCA) from MD simulation done by GROMACS [1].
![[Tutorial] Installing VIAMD on Ubuntu (Linux).](https://img.bioinformaticsreview.com/uploads/2023/12/01184130/viamd.jpg)
Visual Interactive Analysis of Molecular Dynamics (VIAMD) is a tool that allows the interactive analysis of molecular dynamics simulations [1]. In this article, we are installing it on Ubuntu (Linux).

In this tutorial, we will perform energy minimization and equilibration of a simple protein using NAMD [1] & VMD [2]. We are using insulin (PDB ID: 2wfu) for this tutorial.

In this article, we will generate the topology of a small molecule for AMBER forcefield to be used in MD simulation using GROMACS.

Visualizing plots of molecular dynamics simulation is easy once you have generated them. Previously, we provided a few articles on MD output analysis (check the Further Reading section). This article explains how you can easily visualize the plots generated from GROMACS output. (more…)
MD Simulation
How to solve ‘Could NOT find CUDA: Found unsuitable version “10.1”‘ error during GROMACS installation?

Compiling GROMACS [1] with GPU can be trivial. Previously, we have provided a few articles on the same. In this article, we will solve an error frequently occurring during GROMACS installation.

While doing molecular dynamics (MD) simulation, it can be difficult to calculate the number of steps or nsteps for an MD run accurately. In this article, we will learn to calculate nsteps for an MD run. (more…)

We have provided a few articles on GROMACS installation on Ubuntu. In this article, we are going to install GROMACS [1] on Mac OS. (more…)

It is important to see the behavior of protein during an MD simulation. This can be achieved by taking snapshots in the form of PDB format. In this article, we have provided a few commands that you can use to take snapshots of a complete system or protein during MD simulation. (more…)

GROMACS stands for GROningen MAchine for Chemical Simulations [1]. It is a very popular and one of the most widely used open-source bioinformatics software. It is generally used for molecular dynamics simulation of macromolecules. In this article, we will explain its uses and applications in bioinformatics studies. (more…)

We have provided several articles on GROMACS [1] installation on Ubuntu including the easy installation method for GROMACS version 5.x.x. In this article, we will provide shell scripts to install the latest (2021 series) of GROMACS on Ubuntu 18.04 and 20.04. (more…)

MD simulation is a tricky technique if you don’t understand what you are doing through various parameters and algorithms in GROMACS [1]. That may lead to several errors. In this article, we are going to create an index file for the protein groups in GROMACS to solve such errors. (more…)

Generating the topology of small molecules/ligands is an important step in molecular dynamics (MD) Simulation. We explained it in previous articles as part of MD simulation tutorials. In this article, we will explain how can you generate the topology of ligands for MD simulation of complex or small molecules only. (more…)

GROMACS [1] offers a vast range of functions when it comes to molecular dynamics simulation. Today, we are going to explore it for the simulation of small organic molecules. (more…)

In this tutorial, we are performing MD simulation in mixed solvents of methanol and water using GROMACS [1,2]. You can follow our previous articles for MD simulation of a simple protein and a protein-ligand complex. (more…)

We have provided several tutorials on molecular dynamics (MD) simulation (please check further reading section). They include installation of simulation software, simulation of a simple protein, and a complex. In this article, we will analyze the GROMACS [1] output of MD simulation of a complex. (more…)

Molecular dynamics (MD) simulation is one of the most widely used methods in bioinformatics. It needs high computation time and therefore, performed on workstations and servers. It requires software to upload and download files to and from the server. In this article, we have explained how to submit MD simulation jobs on cluster computers using PBS scripts. (more…)

GROMACS [1] is one of the most popular software in bioinformatics for molecular dynamic (MD) studies of macromolecules. We have provided different tutorials regarding MD simulation using GROMACS including its installation on Ubuntu. In this article, we will install GROMACS with GPU acceleration. (more…)

GROMOS96 is a well-known software package used for biomolecular simulations [1]. It can be used for the molecular dynamics simulation of protein, peptide, and protein-ligand complex as well. In this article, we will install GROMOS96 on Ubuntu. (more…)
Molecular dynamics (MD) simulation is considered amongst the important methods in bioinformatics. Installation of MD simulation software and execution of their commands is critical. It requires several parameters to be considered before performing simulations. A single mistake may result in impractical outputs. In this article, we will discuss such important things to remember during the MD simulation and installation and execution of its software (GROMACS) [1,2]. (more…)

You must be logged in to post a comment Login