As I have discussed in my earlier articles about the multiple sequence alignment (MSA) tools (MUSCLE & T-COFFEE). Now in this article, we will discuss different aspects of these tools and which one is more preferred over the another. MUSCLE and T-COFFEE both are multiple sequence alignment tools and also helps to study the evolutionary relationships among the species.As I have already explained the algorithms involved in both the tools which are comparable. During the alignment using MUSCLE, it uses the UPGMA tree construction method which assumes that mutation occurs at the constant rate. This may be a fact which makes it different from other tools.
On the positive side, MUSCLE is a tool which is known for its speed and accuracy on each of the four benchmark test sets ( BAliBASE, SABmark, SMART and PREFAB). It is much faster than other MSA tools. MUSCLE also uses a progressive alignment which is iterated while it gets a better SP score (explained in “Basic concept of MSA” article).
T-COFFEE is an improvisation over MUSCLE in the sense that it combines both global and local alignments which provides better results and it also qualifies the four benchmark tests. Second thing which makes it better than other tools is that it uses an optimization method which provides the multiple alignment that best fits in the input library. T-COFFEE also uses progressive alignment strategy similar to MUSCLE, but unlike MUSCLE, T-COFFEE uses Neighbor Joining tree construction method during alignment which corrects the assumption of UPGMA method and assumes that mutation never occurs at a constant rate.
Let us take protein sequences of ‘Keratin’ protein of few species and align them using both the tools and construct the respective phylogeny trees. In this example, I have taken FASTA sequences of: Homo sapiens (GI: 7717238) , Paralichthys olivaceus (GI: 10716084), Pseudomonas viridiflava (GI: 934022154) and Pseudomonas aeruginosa (GI: 856785229). The results are as follows:
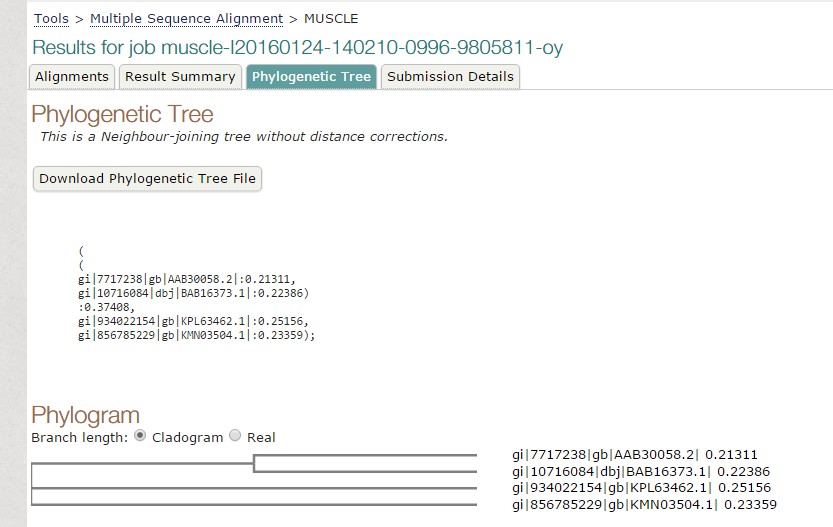
Fig 1. Tree constructed using MUSCLE.
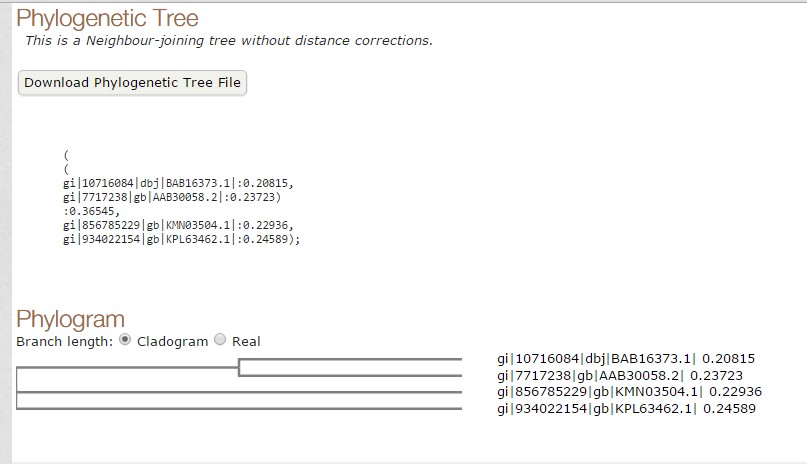
Fig 2. Tree constructed using T-COFFEE.
As we have seen both the trees are slight different. The sequence of Paralichthys olivaceus is placed below to that of Homo sapiens, but it is placed above in tree constructed by T-COFFEE. Similarly, this is case with other two species. This is how MUSCLE & T-COFFEE are different from each other. T-COFFEE is more preferred over MUSCLE while aligning both closely or distantly related species but MUSCLE ia more suitable to align distantly related species since it uses global alignment only, but T-COFFEE uses both.
Note:
An exhaustive list of references for this article is available with the author and is available on personal request, for more details write to muniba@bioinformaticsreview.com.