In my last article I discussed about the Multiple Sequence Alignment and its creation. Now in this article, I am going to explain the workflow of one of the MSA tool, i.e., MUSCLE. MUSCLE is a software which is used to create MSA of the sequences of interest. MUSCLE is one of the softwares which is known for its speed and accuracy on each of the four benchmark test sets ( BAliBASE, SABmark, SMART and PREFAB). It is comparable to T-COFFEE & MAFFT (these tools will be explained in upcoming articles).
MUSCLE algorithm:
Two distance measures are used by MUSCLE for a pair of sequences: a kmer distance (for an unaligned pair) and the Kimura distance (for an aligned pair). A kmer is a contiguous subsequence of length k, also known as a word size or k-tuple, i.e., it decides that how much alphabets in the sequences will be searched & aligned together. Kimura distance is the measure which is based on the fact that multiple substitutions occurs at a single site.
For an aligned pair of sequences, MUSCLE
- computes the pairwise percent identities ,i.e., how much percentage of the sequences are aligned/matched and convert it to a distance matrix applying Kimura.
- Distance matrices are then compiled using UPGMA method (i,e., a method of phylogeny tree construction based on the fact that the mutations occur at the constant rate), which gives a TREE1,which is followed by an progressive alignment and forms MSA1,
- compute pairwise percent identities from MSA1 and construct a Kimura Distance MAtrix,
- again apply UPGMA method and forms TREE2
- again followed by a progressive alignment and forms MSA2, and forms tree.
- now from the last obtained tree it delete the edges which results in the formation of two sub trees,
- computes the sub tree profile (align the sub trees)
- and then finally gives an MSA, for which the SP Score is calculated (explained in previous article “Basic Concept of MSA”),
- if the SP Score is better, then only it saves that last obtained MSA as MSA3, otherwise it discard the MSA,
- again repeat from the step 6, and finally gives a clustered MSA.
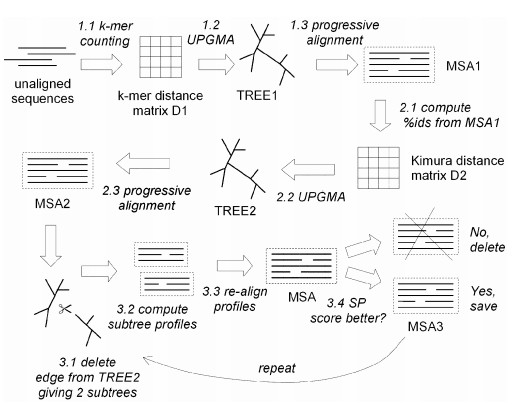
Fig.1 The workflow of MUSCLE
This is how MUSCLE works. MUSCLE alignment is also used in MEGA6 tool which is used for phylogeny tree construction. Every software or tool has its own benefits depending up on the needs under consideration. There are various other tools also available for MSA such as T-COFFEE, MAFFT, etc, which have high accuracy and speed. They will be explained in upcoming articles.
Reference:
MUSCLE : multiple sequence alignment with high accuracy and high throughput
Robert C. Edgar*