If you want to get a quick idea about the non-synonymous vs synonymous (dN/dS) substitutions, you can easily use MEGA software [1]. Although HYPHY/Datamonkey provides the best results regarding selection pressure analyses. MEGA also uses HYPHY program [2] to calculate the dN/dS substitutions rate. Here is how you can do it.
You will need a codon fasta file genes, if you have protein sequences, then convert them into nucleotide codon sequences.
i) open MEGA --> Align --> Edit/Build Alignment --> Retrieve sequences from a file. (If you already have an alignment file then skip this step).
ii) Edit --> Select all --> Align by ClustalW/Muscle.
iii) Save the session and export alignment in MEGA format (.meg).
iv) Minimize the alignment window. Go to the main window and click on "Selection" --> "Estimate selection for each codon (Hyphy)".
v) It will prompt to select a .meg file, select it.
vi) It will ask for analysis preferences as shown in Fig. 1. Choose according to your requirements and click "Compute".
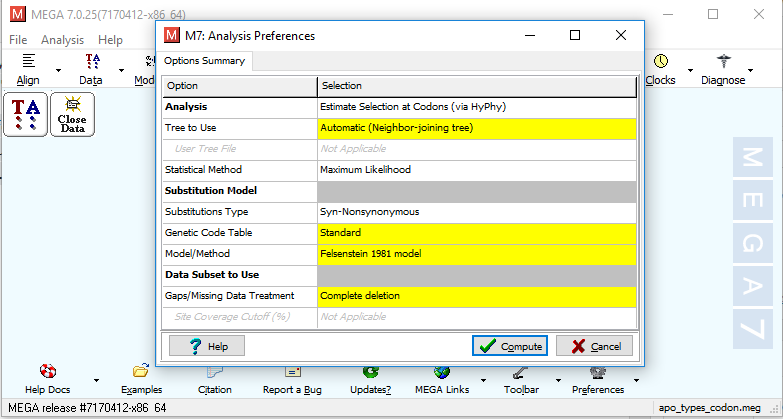
Fig. 1 Analysis preference window for dN/dS substitution calculation using MEGA7.
vii) Later, it will ask for the output format and output directory where you want to save the results. Click "Ok". Your job will be finished after a few minutes depending up on the number and length of sequences.
References
- Kumar, S., Stecher, G., & Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular biology and evolution, 33(7), 1870-1874.
- Pond, S. L. K., & Muse, S. V. (2005). HyPhy: hypothesis testing using phylogenies. In Statistical methods in molecular evolution (pp. 125-181). Springer, New York, NY.




![[Tutorial] Installing Autodock-Vina-develop package on Ubuntu.](https://bioinformaticsreview.com/wp-content/uploads/2021/10/adv-dev.jpg)

