
MAIRA is a new software for taxonomic and functional analysis of long reads without requiring any external resource [1]. It can easily facilitate the execution of metagenomic analysis pipelines on a local computer. It is fast and allows users to perform several analyses in real-time including genus-level analysis and frame-shift alignment of DNA reads.
The main advantage of this software is that it can be easily run on a laptop without any external source support, especially when we have to use workstations for large datasets.
How does MAIRA work?
The complete workflow of MAIRA is shown in Figure 1.
- At first, long reads obtained from a sequencing device are aligned at the protein level against a precomputed database of genus-specific marker genes.
- Based on this alignment, the genus profile is made in real-time.
- The user triggers the presence of specific genera which marks the ‘On-demand species-level analysis’.
- The protein alignment obtained at the second step is aligned against a full database of proteins associated with the triggered genus.
- Finally, a protein graph is generated including the details about the identification of species, resistance, and virulence factors.
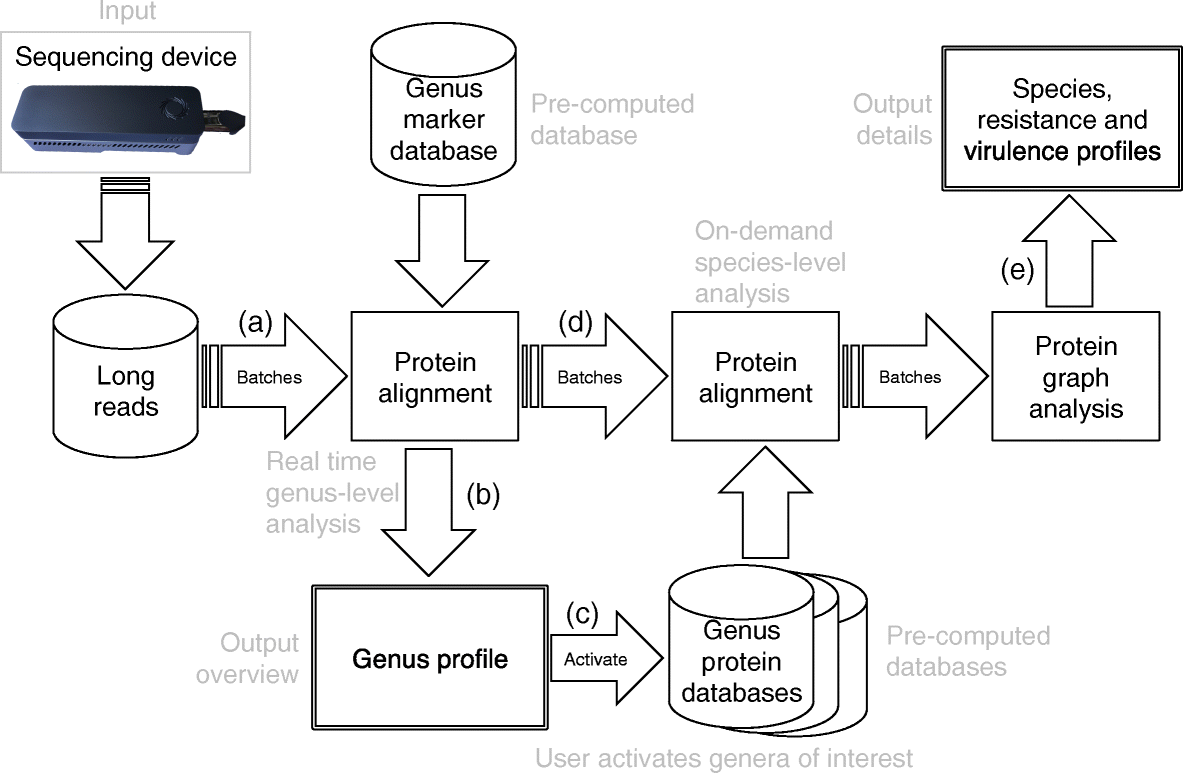
Figure 1 The complete workflow of MAIRA (Figure source: Albrecht et al., 2020) [1].
The software can be downloaded from here.
For further details about MAIRA, read here.
References
- Albrecht, B., Bağcı, C., & Huson, D. H. (2020). MAIRA-real-time taxonomic and functional analysis of long reads on a laptop. BMC bioinformatics, 21(13), 1-12.







