High throughput sequencing is most widely used as it saves a lot of time and provide good results, and produces a huge amount of data which is difficult to manage and especially the tasks and operations performed on it are also very difficult. To ease this purpose, a Python framework have been introduced by Simon Anders and team members, this framework is known as “HTSeq”.HTSeq is a Python library which easily develops the scripts required to fulfill a particular task on the HT data. Basically,HTSeq reads various formats and break it down into recognized strings of characters for further analysis. It also consists of different classes genomic coordinates, sequences, sequencing reads, alignments, gene model information, etc.
Two stand-alone applications have also been developed along with HTSeq, namely, htseq-qa for read quality assessment and htseq-count for preprocessing RNA-Seq alignments for analyzing differential expression.
HTSeq can read various formats such as FASTA, FASTQ (short reads), SAM/BAM (short-read alignments). Wherever appropriate, different parsers will yield the same type of record objects. For example, the record class SequenceWithQualities is used whenever sequencing read with base-call qualities needs to be presented, and hence yielded by the FastqParser class and also present as a field in the SAM_Alignment objects yielded by SAM_Reader or BAM_Reader parser objects (Fig. 1). There are some specific classes to represent Genomic Position and Genomic Intervals of the sequence. In order to achieve good performance, various parts of HTSeq is written in ‘Cython’ ( a tool which translates Python code augmented with C).
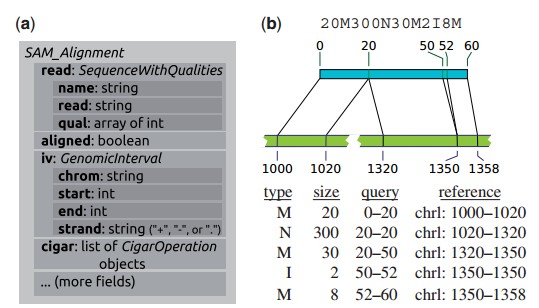
Fig. 1. ( a) The SAM_Alignment class as an example of an HTSeq data record: subsets of the content are bundled in object-valued fields, using classes (here SequenceWithQualities and GenomicInterval) that are also used in other data records to provide a common view on diverse data types. ( b) The cigar field in a SAM_alignment object presents the detailed structure of a read alignment as a list of CigarOperation. This allows for convenient downstream processing of complicated alignment structures, such as the one given by the cigar string on top and illustrated in the middle. Five CigarOperation objects, with slots for the columns of the table (bottom) provide the data from the cigar string, along with the inferred coordinates of the affected regions in read (‘query’) and reference.
HTSeq also consists of a class which deals with the gapped alignments, namely SAM_Alignment, with multiple alignments and with paired-end data. HTSeq provides a function, pair_SAM_alignments_with_buffer, to pair up the alignment records by keeping a buffer of reads whose end pair has not yet been found, and so facilitates processing data on the level of sequenced fragments rather than reads. HTSeq also facilitates the storage of genome-position-dependent data, which means that each base pair position on the genome can be given a particular value that can be easily stored and retrieved by simply entering the same value.
The script htseq-qa is a simple tool for initial quality assessment of sequencing runs. It produces plots that summarize the nucleotide compositions of the positions in the read and the base-call qualities. As we discussed earlier in this article that htseq-count is a tool for RNA-Seq data analysis. It counts for each gene that how many aligned reads overlap the sequence exons. Since it is designed specifically to analyse differential expression only reads mapping unambiguously to a single gene are considered and the reads aligned to multiple positions or overlapping with more than one gene are discarded. In case of paired-end data, htseq-count counts only the fragment not the reads because the two paired ends originating from the same fragment provide only evidence for one cDNA fragment and should hence be counted only once.
In this way, HTSeq offers a comprehensive solution to facilitate a wide range of programming tasks in HTS data analysis. For further reading, click here.
Note:
An exhaustive list of references for this article is available with the author and is available on personal request, for more details write to muniba@bioinformaticsreview.com








Very nice article. Nicely Written and very informative.
Thank you sir.
You should think about compiling all the sequencing format types used in Bioinformatics in the form of an article..the title can be “Sequencing Formats you should be aware of: the variety they offer in usage”. This will be very good for beginners and graduate students. After this, the second article on the same line should be “How to generate these formats using online resources”. Think about it. It would be very informative and useful for users…Cheers
Ok sure sir, i will do next time