What is predictive metagenomics profiling?
Recently, predictive metagenomics profiling (PMP) has been added to the microbial ecologist’s arsenal of strategies for probing microbial communities. Currently, there are two platforms available for PMP; PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States; http://picrust.github.io/picrust/) and tax4fun available at tax4fun.gobics.de/ (Langille et al., 2013; Aßhauer et al., 2015). The aim of PMP is to predict the abundance of functional gene families present in microbial communities (i.e. the community’s functional potential) using amplicon-based sequencing data, such as 16S rRNA data.
If PMP reveals that the predicted functional potential of a community changes between treatments, then researchers will have a strong incentive to invest the time, money and computational power to investigate community function further via techniques including shotgun metagenomics or functional microarrays such as GeoChip (He et al., 2010). Additionally, the types of gene families predicted to vary by PMP could give rise to new hypotheses that will direct future experimental design.
How does it work?
PICRUSt and tax4fun are described in detail in their respective publications but briefly:
Both programs generate a database of organisms with known gene content. For tax4fun, this is achieved using KEGG organisms (Kyoto Encyclopedia of Genes and Genomes; www.genome.jp/kegg/) with sequenced genomes. For PICRUSt, a reference phylogenetic tree with GreenGenes identifiers (available at greengenes.lbl.gov/) is created. The gene content for organisms in the PICRUSt reference phylogenetic tree is either a) compiled directly from databases containing the well annotated, sequenced genomes or, b) inferred using an ancestral state reconstruction algorithm. The latter process is used when organisms in the reference phylogenetic tree have not been sequenced. The underlying assumption is that taxonomically similar organisms will have functionally similar capabilities. Indeed, the authors of PICRUSt highlight that phylogeny and biomolecular function are highly correlated (Langille et al., 2013).
Gene family predictions for experimental data then are made by associating the taxonomic ID of OTUs (Operational Taxonomic Units) in experimental data to organisms in the reference database with precomputed gene content. For PICRUSt, GreenGenes taxonomic identifiers are used to create the phylogenetic reference tree, thus to map the experimental 16S rRNA data to this tree, taxonomic assignments of OTUs must also be performed using GreenGenes. Conversely, tax4fun requires SILVA (www.arb-silva.de) assigned taxonomies which are then mapped to the reference database of KEGG organisms. Both programs use 16S rRNA copy number information to normalize the abundance of identified OTUs.
Why use predictive metagenomics profiling?
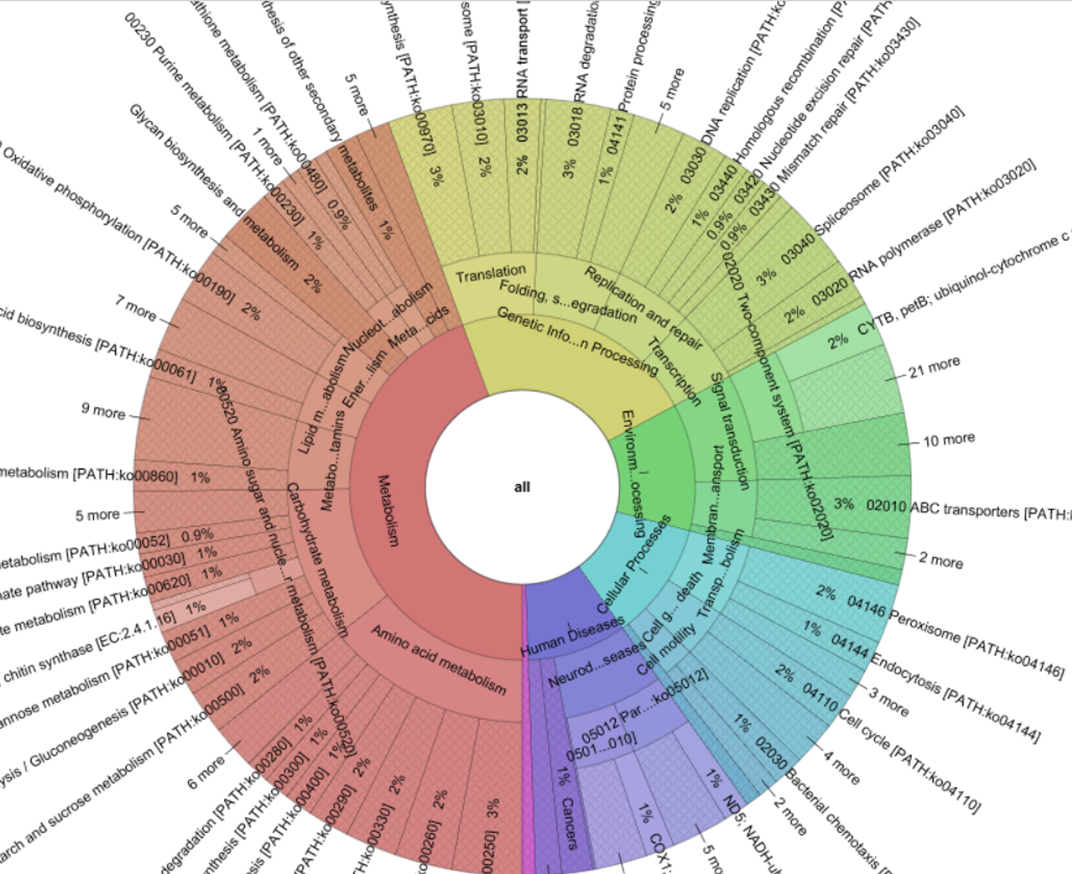
Unlike metagenomics, which requires massive amounts of sequencing in order to achieve adequate read-coverage of rare genomes – which is particularly challenging for highly diverse communities such as soils – PMP only requires sufficient sequencing depth to cover a single target gene – i.e. the 16S rRNA gene (Howe et al., 2014). This significantly reduces the amount of data required to take a first look at the community functional profile. The output of a PMP pipeline is a table of predicted gene family counts (in the form of identifiers such as KEGG ontologies) which can be clustered to path-way level categories (i.e. KO modules) if so desired. Additionally, for each OTU, 16S rRNA copy number is used to scale the contribution of predicted gene families to the community’s functional potential.
A natural limitation for PMP lies in the diversity of available reference organisms that have their genomes fully sequenced. For an OTU to have its gene content predicted it must be able to be mapped to a reference organism. If it is not similar enough to a reference organism then gene content cannot be predicted. Additionally, even though PICRUSt can predict the gene content of unsequenced organisms, these predictions will only be accurate if there are a number of closely-related, sequenced genomes available to make predictions from. Nevertheless, PMP is a simple and elegant strategy for adding value to 16S rRNA metagenomic data, such as the one that would be generated on an Illumina Miseq.
Using predictive metagenomics profiling
The input for PMP is a normalized OTU table whereby taxonomic assignments have been made using the database appropriate to the PMP strategy (i.e. SILVA or GreenGenes). As with any sequencing data, appropriate QC and data trimming strategies need to be observed. Data QC, taxonomic assignments, generation of an OTU table and normalisation (i.e. rarefied counts or model-based normalisation) can be performed using existing pipelines such as QIIME (Quantitative Insights Into Microbial ecology; www.qiime.org) and USEARCH 16S rRNA pipelines (Caporaso et al., 2010; Edgar, 2010; McMurdie and Holmes, 2014). Both PICRUSt and tax4fun are compatible with QIIME outputs, making it easy for users to add PMP to existing workflows. Through the QIIME-PICRUSt help pages, there is a range of recommended scripts that can be used to assess changes to functional profiles between communities (Caporaso et al., 2010). Additionally, KO data can be exported and used in conjunction with traditional ecological tools, such as ordinations (NMDS, PCO etc.,) or species indicator tests (available using the multipatt function in the indicspecies package in R) to identify gene families or functional modules that are indicative of different communities (De Cáceres et al., 2010).
Thus, given a 16S rRNA dataset, PMP is a cost-effective, a logical starting point for deciding a) whether to proceed with an examination of functional changes in a community and b) the best experimental strategy to investigate said community.
References
Aßhauer, K.P., Wemheuer, B., Daniel, R., Meinicke, P., 2015. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics, 31, 2882-2884. Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K., Fierer, N., Pẽa, A.G., Goodrich, J.K., Gordon, J.I., Huttley, G.A., Kelley, S.T., Knights, D., Koenig, J.E., Ley, R.E., Lozupone, C.A., McDonald, D., Muegge, B.D., Pirrung, M., Reeder, J., Sevinsky, J.R., Turnbaugh, P.J., Walters, W.A., Widmann, J., Yatsunenko, T., Zaneveld, J., Knight, R., 2010. QIIME allows analysis of high-throughput community sequencing data. Nature Methods, 7, 335-336. De Cáceres, M., Legendre, P., Moretti, M., 2010. Improving indicator species analysis by combining groups of sites. Oikos, 119, 1674-1684. Edgar, R.C., 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 26, 2460-2461. He, Z., Deng, Y., Van Nostrand, J.D., Tu, Q., Xu, M., Hemme, C.L., Li, X., Wu, L., Gentry, T.J., Yin, Y., Liebich, J., Hazen, T.C., Zhou, J., 2010. GeoChip 3.0 as a high-throughput tool for analyzing microbial community composition, structure and functional activity. ISME Journal, 4, 1167-1179. Howe, A.C., Jansson, J.K., Malfatti, S.A., Tringe, S.G., Tiedje, J.M., Brown, C.T., 2014. Tackling soil diversity with the assembly of large, complex metagenomes. Proceedings of the National Academy of Sciences of the United States of America, 111, 4904-4909. Langille, M.G.I., Zaneveld, J., Caporaso, J.G., McDonald, D., Knights, D., Reyes, J.A., Clemente, J.C., Burkepile, D.E., Vega Thurber, R.L., Knight, R., Beiko, R.G., Huttenhower, C., 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 31, 814-821. McMurdie, P.J., Holmes, S., 2014. Waste Not, Want Not: Why rarefying microbiome data Is inadmissible. PLoS Computational Biology, 10.
For a more comprehensive list of references, readers are directed to mail us at info@bioinformaticsreview.com