In order to study gene regulation, it is necessary to identify the target sites of miRNA in mRNA. miRNAs have been the main point of research as its binding to mRNA degrades the target mRNA and also prevents the translation of target mRNAs [1-4]. The identification of miRNA target sites, target mRNAs and the potential functional roles of miRNA may be assigned.
There are seven features commonly used to predict the miRNA target sites (described in previous article Common features used to develop miRNA target prediction tools), and these methods are considered to be conventional to predict the target sites of miRNA. There are many target prediction tools available which are based on the seven conventional features of target prediction such as miRANDA uses seed match, free energy, and conservation [5], TargetScan utilizes seed match, pairing of mRNAs with 3′ of miRNAs, local AU content, etc., [6,7], and so on. Recently, few new features have been developed by Ding et al., (2016), they have applied four different machine learning approaches on the CLASH data [8]. CLASH (Crosslinking Ligation And Sequencing of Hybrids) is an experimental procedure, which implements a high-throughput approach to identify the sites of RNA- RNA interaction [9]. CLASH method is optimized to study the miRNA targets using Argonaute proteins [9]. Despite other high-throughput experimental methods such as PAR-CLIP [10,11], and HITS-CLIP [12], the CLASH experiments provide a better understanding of miRNA target sites and help to develop better computational methods for miRNA target prediction.
Ding et al., (2016) has developed a random forest approach known as TarPmiR (http://hulab.ucf.edu/research/projects/miRNA/TarPmiR/), in which they have incorporated six conventional features and seven new features for miRNA target site prediction [8]. According to Ding et al., (2016), the newly incorporated features are:
- m/e motif
It is the pairing probability of miRNA. If miRNA residues at positions in seed regions match the residues at the corresponding positions in target sites, then it is marked as ‘m’ (match) and if they do not match, and tends to form mismatches or bulges, then it is marked as ‘e’ (else). The probability score of m/e at each position of miRNAs is calculated as:
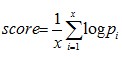
where x is the length of the miRNA which is smaller than 24.
- Total number of paired positions
The total number of paired positions for each miRNA-mRNA binding site is calculated.
- Target mRNA region length
It is the length of the number of residues of miRNA exactly binding to the target mRNA.
- Largest consecutive pairs length
It is calculated as the length of the largest consecutive pairs to the 5’ end of miRNA.
- Length of the largest consecutive pairs allowing 2 mismatches
This feature allows 2 mismatches in the relative position of largest consecutive pairs to the 5′ of miRNA.
- Largest consecutive pairs position
It is calculated as the relative position of largest consecutive pairs to the 5’ end of miRNA.
- Position of the largest consecutive pairs allowing 2 mismatches
This feature is similar to the largest consecutive pairs position and is calculated as the relative position of largest consecutive pairs to the 5′ end of miRNA allowing 2 mismatches.
- Exon Preference
It considers the preference of miRNA- mRNA binding in terms of an exon. If miRNA binds to a specific exon then, this feature assigns a score, otherwise, it remains zero.
- Difference between the number of paired positions in the seed region and in the 3′ end of miRNA
This feature counts the difference between the number of paired residues in the seed region and that within the 3′ end of miRNA.
TarPmiR is developed including all the conventional and the new features [8]. This tool has been proved more efficient than the other available tools and found to provide less number of false positives and true negatives [8]. It is also available Linux (http://hulab.ucf.edu/research/projects/miRNA/TarPmiR/). For further reading please click here.
References:
1. Axtell,M.J. et al. (2011) Vive la difference: biogenesis and evolution of microRNAs in plants and animals. Genome Biol., 12, 221.
2. Bartel,D.P. (2009) MicroRNAs: target recognition and regulatory functions. Cell, 136, 215–233.
3. Muljo,S.A. et al. (2010) MicroRNA targeting in mammalian genomes: genes and mechanisms. Wiley Interdisc. Rev. Syst. Biol. Med., 2, 148–161
4. Wang,Y. et al. (2011) Transcriptional regulation of co-expressed microRNA target genes. Genomics, 98, 445–452.
5. Enright,A.J. et al. (2004) MicroRNA targets in Drosophila. Genome Biol., 5, R1-R1.
6. Friedman,R.C. et al. (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res., 19, 92–105.
7. Grimson,A. et al. (2007) MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell, 27, 91–105.
8. Ding J. et al., (2016). TarPmiR: a new approach for microRNA target site prediction. Bioinformatics, 32(18), 2016, 2768–2775 doi: 10.1093/bioinformatics/btw318
9. Helwak, A. et al. (2013) Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell, 153, 654–665
10. Chi,S.W. et al. (2009) Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps. Nature, 460, 479–486.
11. Licatalosi,D.D. et al. (2008) HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature, 456, 464–469.
12. Hafner,M. et al. (2010) Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell, 141, 129–141.







