Previously, we have explained the initial methods involved in the structure prediction of a protein. In that article, we discussed the three basic methods involved in protein structure prediction: Homology modeling, ab-initio, and threading. In this article, we will explain the methodology involved in performing the homology modeling of a simple protein.
Homology modeling uses a template structure to predict a new structure for a query protein. We go for homology modeling when enough similarity (~ 30-32 %) for the query protein can be found in the available databases. The most widely used and accurate prediction server is the Swiss Model [1]. A complete flowchart is shown below in Figure 1.
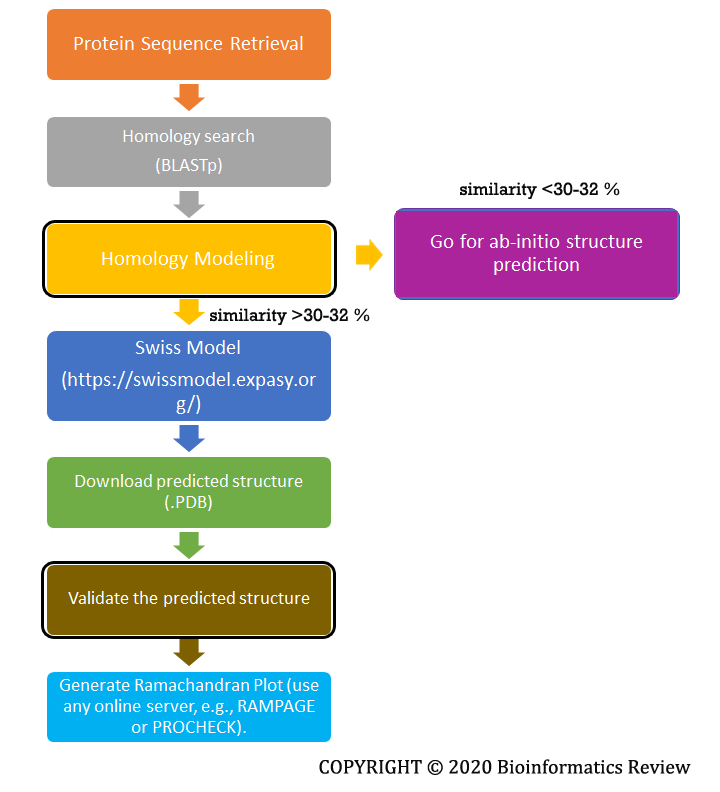
Figure 1 Basic steps involved in homology modeling.
These are the basic steps followed during the homology modeling of a simple protein. There are several other software/tools that are used for homology modeling but the Swiss Model provides the most accurate results. Ab-initio prediction will be explained in upcoming articles.
References
- Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., … & Lepore, R. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic acids research, 46(W1), W296-W303.