GROMACS stands for GROningen MAchine for Chemical Simulations [1]. It is a very popular and one of the most widely used open-source bioinformatics software. It is generally used for molecular dynamics simulation of macromolecules. In this article, we will explain its uses and applications in bioinformatics studies.
Contents
Here are some of the primary uses of the GROMACS software:
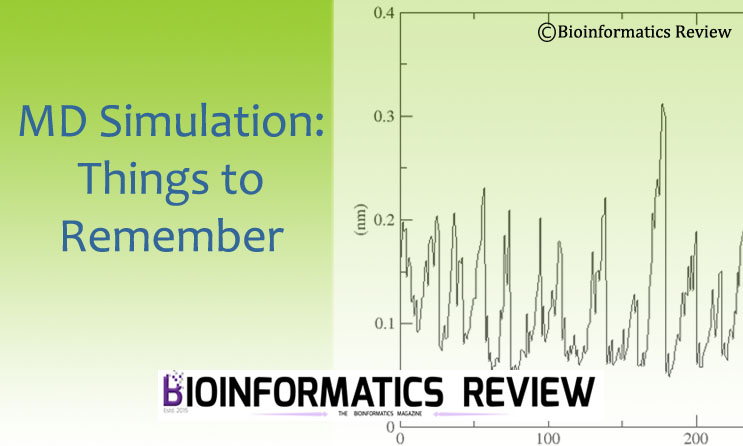
- GROMACS is used to perform molecular dynamics simulations of biological macromolecules such as proteins, nucleic acids, and lipids, and also for non-biological systems such as polymers. GROMACS is very fast and efficient in calculating the non-bonding interactions.
- It helps to study the behavior of systems (for example, protein-ligand complexes) under a specific temperature, pressure, and pH.
- GROMACS also consists of a set of tools for preparation and analysis including free-energy calculation.
- It helps in monitoring the physical movements of atoms and molecules at specific time intervals.
- It can be used to analyze the different conformations of ligands lying inside the binding pocket while keeping the other amino acids or molecules fixed at a particular position.
- GROMACS has different force fields that can be used for energy minimization and different kinds of systems.
- Simulation of bonding interactions between two protein structures can be easily studied using GROMACS.
References
- Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., & Lindahl, E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1, 19-25.
Further Reading
Tutorial: MD simulation output analysis of protein using GROMACS
Tutorial: MD simulation output analysis of a complex using GROMACS
Method-2: Installing GROMACS on Ubuntu 20.04 with CUDA GPU Support