The interaction amongst plasma proteins and drugs is an imperative pharmacological parameter which impacts their pharmaco-dynamic behaviors and influences the circulation and disposal of a drug, the structure, and physiological activity of transporter proteins [1, 2]. The proteins bind to various drugs and the most essential being albumin. A drug’s proficiency might be influenced by how much it binds to the proteins in blood plasma[3]. The less bound a drug is, the all the more effectively it can cross cell layers or diffuse. Protein binding can impact the drug’s organic half-life in the body[4]. The bound segment may go about as a repository or terminal from which the drug is gradually discharged as the unbound form. Since the unbound form is being metabolized and additionally discharged from the body, the bound portion will be discharged in order to maintain equilibrium. Since albumin is alkalotic, acidic and neutral drugs will principally bind to albumin [5].
Generally, for drugs bound under 90%, the extent of the impact is so little as to be unimportant. Interestingly, drugs bound over 90% might be vitally influenced by the modifications in the binding. As an instance NSAIDs (Non-steroidal Anti-inflammatory Drugs) are over 99% bound to albumin. Thus, less than 1% is free in plasma and constitutes the dynamic moiety [6]. In uremia, what may have all the earmarks of being an inconsequential reduction in binding from 99% to 98% really brings about a multiplying of the free unbound concentration from 1-3% – a greatness of progress that might be clinically essential [7]. Interestingly, just half of methotrexate is bound to albumin. A lessening in binding to 45% would have just a minor, clinically unimportant effect on unbound methotrexate concentrations [8].
A successive misguided judgment is that such an impact brings about expanded concentrations of the unbound pharmacologically dynamic drug, creating an improved impact—including toxicity. In the larger part of occasions, however, there is no expansion in the concentration of the unbound drug and along these lines no alteration in response unless brought on by different variables.
Human Serum Albumin
HSA is the most abundant protein found in human blood. It has an imperative part in keeping up the colloidal osmotic weight in blood; it transports and appropriates exogenous and endogenous particles and metabolites, for example, nutrients, hormones, unsaturated fats, and numerous differing drugs [9, 10].
Structure of HSA
This heart-formed protein which has a net charge of −15, at impartial pH, and an isoelectric point of around 5, it comprises of 585 amino acids deposits and is made out of three basically similar domain sites (I, II, and III). Each of the domains contains two subdomains (A and B) and is balanced out by 17 disulfide bridges [11-13]. HSA has binding sites for heterocyclic ligands and aromatic ligands inside two hydrophobic pockets which are unraveled by the crystal structure analysis. In subdomains IIA is usually alluded to as Sudlow’s site I have also known as warfarin-binding site and IIIA is regularly alluded to as Sudlow’s site II known as indole/benzodiazepine site[14]. Both hydrophobic and electrostatic collaborations assume a noteworthy part in controlling the affinity towards drug binding for sites I and II; for the site I, for the most part, hydrophobic connections are prevailing, while for site II, a blend of hydrophobic, hydrogen holding, and electrostatic associations all assume an urgent part [15].
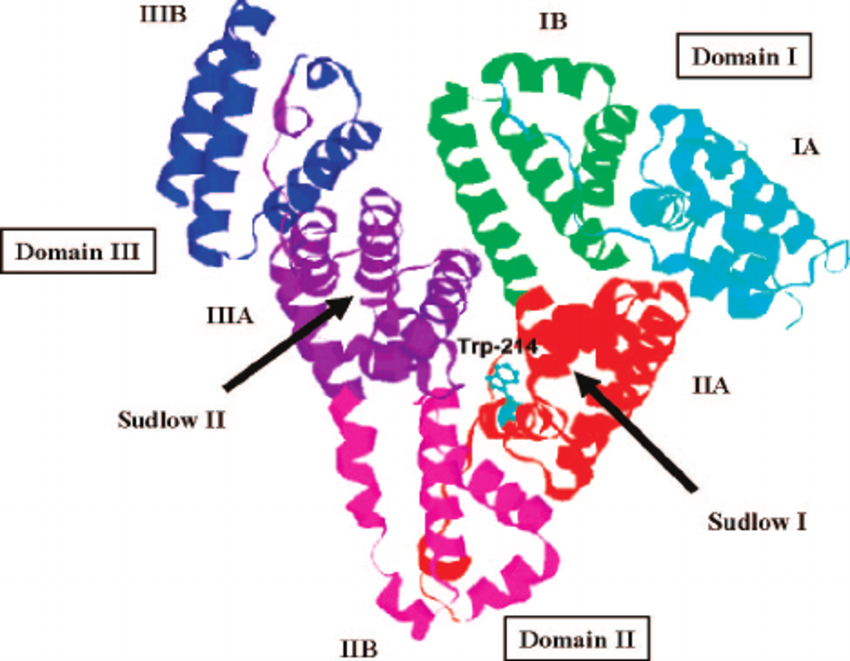
Figure 1 Crystal structure of HSA and the locations of domain-binding sites. The locations of hydrophobic binding sites (Sudlow I and Sudlow II) are indicated. The position of the tryptophan residue (Trp-214) in the middle of helix H2 in subdomain II. [16]
Various Binding Sites of HSA for Fatty Acids
HSA can bind seven counterparts of long-chain unsaturated fats (FAs) at various binding sites with various affinities. In sites FA1 – 5 the carboxylate moiety of unsaturated fats is anchored by electrostatic/polar associations.
Despite what might be expected, sites FA6 and FA7 don’t show an unmistakable confirmation of polar associations that keep setting up the carboxylate leader of the unsaturated fat, hence proposing that sites FA6 and FA7 are low-affinity FA binding sites.[17]
Table1 Binding sites of Human Serum Albumin for Fatty acids [17].

Figure 2 Three-dimensional structure of HSA complexed with endogenous and exogenous ligands bound to the FA sites. The subdomains of HSA are rendered with different colors (domain IA, in blue; domain IB, in cyan; domain IIA, in forest green; domain IIA, in green domain IIIA, in yellow; domain IIIB, in orange)[18]. The picture has been drawn with the UCSF Chimera package [19, 20].
CMPF
3-Carboxy-4-methyl-5-propyl-2-furanpropionic acid (CMPF) is additionally called 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid and 2-(2-carboxyethyl) – 4-methyl-4-propylfuran-3-carboxylic acid. CMPF is essentially gathered in the serum of patients with interminable kidney sickness and is thought to be a strong uremic toxin [21]. CMPF was initially recognized in the urine of humans in 1979 [22] and it is accepted to be produced from the utilization of fish, vegetables and organic products [23]. Be that as it may, it can’t be barred that a segment of CMPF is formed de novo in the human body [24]. CMPF is a solid inhibitor of mitochondrial respiration [25]. CMPF is also related to dysfunction of the thyroid gland. CMPF likewise straightforwardly hinders renal discharge of different drugs and endogenous organic acids by intensely repressing OAT3 transporters [26]. It is additionally thought to add to different neurological variations from the norm since it hinders the transport of organic acids at the blood-brain barrier [22] and erythropoiesis [27] under CKD conditions.
Structure of CMPF
CMPF, one of the significant UTs, has a furan ring and two carboxylic acid moieties in its structure (Fig. 2) [28]. CMPF amasses at unusually abnormal states in the serum of uremic patients because of a diminishment of renal clearance, and its serum levels (400 mM) have been accounted for to be 10-overlay higher than that under typical physiological conditions (40 mM) [29].
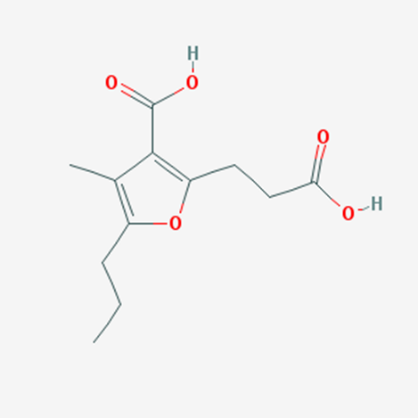
Figure 3 Chemical structure of CMPF (Source: PubChem)
Ligand Binding to HSA
HSA, the most unambiguous protein in plasma, is a standout amongst the most widely researched proteins. HSA is integrated into the liver and traded as a solitary non-glycosylated chain, achieving a blood grouping of around 7.06 1074 M. HSA is best known for its remarkable ligand binding limit, giving an end to a wide range of aggravating that might be accessible in amounts well past their solvency in plasma. Under physiological conditions, HSA typically binds up to two moles of unesterified fatty acids, in spite of the fact that it might suit up to six moles under certain disease states.
Conformational Changes in HSA after Ligand Binding
Strikingly, HSA experiences pH-and allosteric impact or dependent reversible conformational isomerization(s) HSA demonstrates a quick (F) form, described by an increment in viscosity, low solubility, and a critical loss in the a-helical content. HSA shows the neutral (N) form which is portrayed by the heart-shaped structure. Between pH 4.3 and 8.0 within the sight of allosteric effectors (e.g., drugs and long-chain FAs) and at pH more prominent than 8 without ligands, HSA changes adaptation to the fundamental (B) frame with the departure of a-helix and an expanded affinity for a few ligands, similar to warfarin. The hydrophobic bonds between the long FA polymethylenic tail and HSA drive allosteric alteration.
It is entrenched that the proximity of hydrophobic moieties and hydrophilic negatively charged groups are the fundamental auxiliary prerequisites for ligand binding to HSA. The affinity to HSA increments of about one order of magnitude for each hydrophobic moiety included.
Genetic Variations in HSA for Ligand Binding
Almost all the known natural genetic variants of HSA have point mutations that result in a changed charge located on the surface of the HSA molecule and, therefore, it is not likely that the mutated residues directly take place in the formation of the high affinity binding sites The effect of genetic variation on the FAs-binding properties has been investigated using several HSA and pro-HSA mutants and the results showed that also single amino acid substitutions can, quantitatively and/or qualitatively, affect the binding capacity of (pro-)HSA for FAs. A given ligand may display different binding properties depending on the concomitant association or dissociation of different ligand(s) [30].
CMPF Binding to HSA
CMPF which is a furan fatty acid uremic toxin (UT) binds to HSA and affects protein drug binding consequently reducing the serum binding of drugs since HSA is the most important drug carrier protein. Among all the uremic toxins, CMPF is found to have the strongest binding affinity to HSA. CMPF is the most powerful inhibitor and its binding site agreed with that of bilirubin (site 1). Uremic toxins (UTs) accumulate in the body of hemodialysis patients. UTs often exert adverse effects on patients and cause significant interactions with clinically relevant drugs. As per a research, the plasma concentrations of three typical anionic UTs, indoxyl sulfate (IS), 3-indoleacetic acid (IA), and CMPF is assayed in 20 hemodialysis patients and 5 healthy volunteers. The findings state that CMPF concentrations in the plasma of patients were unaltered prior and then afterward dialysis (63.0 ± 6.3 μM and 65.1 ± 6.7 μM, individually), and these qualities were around 5-fold more prominent contrasted with those in healthy volunteers. (63.0 ± 6.3 μM and 65.1 ± 6.7 μM, respectively), and these values were about 5-fold greater compared with those in healthy volunteers. Although dialysis decreased the plasma IS concentration from 157.9 ± 19.9 μM to 103.8 ± 13.3 μM, the value after hemodialysis was still ca. 27-fold greater than that in healthy volunteers. IA concentrations before and after hemodialysis were almost identical to those in healthy volunteers. There are no noteworthy contrasts in the plasma concentrations of the three anionic UTs between female and male patients. The magnitude of protein binding is in the order CMPF>IS>IA, indicating that hemodialysis clearance of these anionic UTs is dependent on their protein binding capacity [31] and CMPF has an association constant around 106 -107 /M for Human albumin [32].
Table2 Binding parameters of uremic toxins on HSA at pH 7.4 and 25°C, where K1, the association constant of CMPF at the site I; K2, the association constant of CMPF at site II [32].
Structure of HSA bound CMPF Complex
This surprising binding limit, which regularly truly impacts pharmacokinetic properties of remedial medications, is encoded in the secondary structure of HSA. In particular, the protein is made out of ~70:30% α-helix: arbitrary loops with no β-sheets, which gives the protein a high level of conformational adaptability [33]. Regularly, HSA ligands are suited fundamentally to one of the two high-affinity sites with typical binding association constants in the scope of 104-106M-1[33, 34]. Conceivable conformational changes on binding of FA atoms to HSA have been seen through MD simulations. High-and low-affinity FA-binding sites on HSA have been distinguished in light of binding free energy calculations. Molecular simulation approaches have extraordinary possibilities to give point by point biophysical bits of knowledge into HSA and also the impacts of the binding of FAs or different ligands to HSA [35].
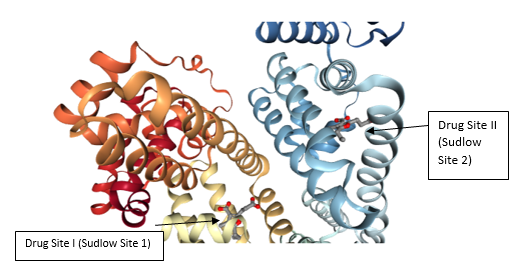
Figure 4 CMPF binding to HSA in two different Binding Sites. Source: RCSB PDB, PDB ID: 2BXA [36].
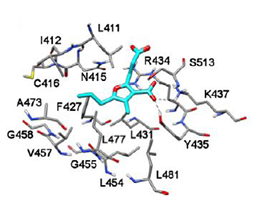
Figure 5(a) Docking of compounds into the site I of HSA: CMPF [38].
According to molecular docking performed using in previous works, at the site I, the 3-carboxylate group of CMPF interacts with Tyr174, His266, and Arg281 while the carboxyethyl group forms H-bonds with Lys223 and Arg246. The propanoic acid group is exposed to the solvent and the n-propyl chain binds into a lipophilic pocket created by Ile412, Phe427, Val457 and Leu477 [38].
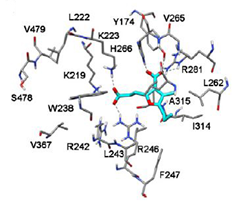
Figure 5(b) Docking of CMPF into the site I of HSA [38].
At site II, the 3-carboxylic group of CMPF interacts with Tyr435 and Lys437 (Fig 5 b) the propanoic acid group is exposed to the solvent and the n-propyl chain binds into a lipophilic pocket created by Ile412, Phe427, Val457 and Leu477 [38].
Predictive Models in Bioinformatics to Study Binding Association & Dissociation Relationships
The previous investigation of the conveyance of percentage plasma protein binding among helpful drugs demonstrated that no broad tenets for protein binding can be inferred, aside from the class of chemotherapeutics, where a reasonable pattern towards lower binding could be watched. For the larger part of signal zones, in any case, exact tenets are absent. In a review, a broad rundown of increase decided essential association constants is made for binding to HSA for 138 compounds from the previous literature where binding constants with the percentage fraction of protein bound are related. Accordingly, it demonstrated that the percentage information over 90%, relating to a binding consistent beneath 6 microM, is of lacking precision. Besides, a non-exclusive model has been made for the forecast of drug association constants to HSA, which utilizes a pharmacophoric similitude idea and Partial Least Square examination (PLS) to develop a quantitative structure-action relationship. It can single out the submicromolar to nanomolar binders, i.e. to separate in the vicinity of 99.0 and 99.99% plasma protein binding [34]. Contingent upon the framework, this can be essential in restorative science programs and may together with other registered physicochemical and ADME properties aid the prioritization of manufactured techniques. Lipophilicity of drugs generally felt to rule binding to HSA, is by all accounts, not the only significant descriptor.
As indicated by another exploration, through superior affinity chromatography the binding affinities to HSA of 95 assorted drugs and comparative mixes have been tentatively decided. This information helped in inferring quantitative structure-action relationship models to anticipate binding affinities to HSA of new compounds on the premise of their structure. Basic straight, one-variable models have been determined for particular groups of compounds (r(2) > or = 0.80; q(2) > or = 0.62): beta-adrenergic foes, steroids, COX inhibitors, and tricyclic antidepressants. Likewise, worldwide models have been determined to be relevant to the entire therapeutic synthetic space by utilizing the full database of HSA binding constants portrayed previously. For this point, a genetic algorithm has been utilized to comprehensively look and select for multivariate and nonlinear conditions, beginning from a vast pool of sub-atomic descriptors. The subsequent models show great fits to the test information (r(2) > or = 0.78; LOF < or = 0.12). Also, both interior (cross approval and randomization) and outside approval tests have shown that these models have great prescient power (q(2) > or = 0.73; PRESS/SSY < or = 0.23; r(2) > or = 0.82 for the outer set) [39]. Factual investigation of the condition populaces demonstrates that hydrophobicity (as measured by the ClogP) is the most imperative variable deciding the binding degree to HSA. Moreover, auxiliary components (particularly the topological (6)chi(ring) file and some Jurs descriptors) likewise every now and again show up as descriptors in the best conditions. It deciphers that binding to HSA ends up being controlled by a mix of hydrophobic forces together with some modulating shape elements.
Conclusion and future developments
CMPF binding with HSA shows highest binding affinity amongst the other uremic toxins, which renders its removal quite difficult using conventional hemodialysis. This results in accumulation of CMPF in the renal cells and ultimately leads to the renal cellular damage. There are many descriptors responsible for the highest binding affinity such as lipophilicity, shape elements etc. Various dialysis methods have been employed for the renal clearance of CMPF, but there is no successful method have been found for the removal of CMPF from the renal cells. This review is focused on highlighting the binding effects of CMPF with HSA. After analyzing the literature, we can conclude that the association and dissociation study of CMPF with HSA is an important concern. Some ways including novel biotechnological assays, computational biology should be explored to study the binding of CMPF with HSA and the factors involved in this association.
References
1. Hu Y-J, Liu Y, Sun T-Q, Bai A-M, Lü J-Q, Pi Z-B. Binding of anti-inflammatory drug cromolyn sodium to bovine serum albumin. International Journal of Biological Macromolecules. 2006; 39(4-5):280–285. PubMed PMID: 16707156
2. Seedher N, Bhatia S. Reversible binding of celecoxib and valdecoxib with human serum albumin using fluorescence spectroscopic technique. Pharmacological Research. 2006;54(2):77–84. PubMed PMID: 16600620
3. Isogai H, Hirayama N. In silico prediction of interactions between site II on human serum albumin and profen drugs. ISRN Pharma. 2013;2013:8 pages.818364. PubMed PMID: 23533820
4. IUPAC,Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”) (1997). Online corrected version: (2006–) “Biological Half Life”.
5. Shargel, Leon (2005).Applied Biopharmaceutics & Pharmacokinetics. New York: McGraw-Hill, Medical Pub. Division.ISBN 0-07-137550-3.
6. Verbeeck RK, Blackburn JL, Loewen GR. Clinical pharmacokinetics of non-steroidal anti inflammatory drugs. ClinPharmacokinet. 1983 Jul-Aug;8(4):297-331. Review. PubMed PMID: 6352138.
7. Keller F, Maiga M, Neumayer HH, Lode H, Distler A. Pharmacokinetic effects of altered plasma protein binding of drugs in renal disease. Eur J Drug MetabPharmacokinet. 1984 Jul-Sep;9(3):275-82. PubMed PMID: 6519129.
8. Rochas MA, Tufenkji AE, Levillain P, Houin G. Protein binding of methotrexate to human albumin and serum. A first derivative spectroscopic analysis. Arzneimittelforschung. 1991 Dec;41(12):1286-8. PubMed PMID: 1815530.
9. Maiti TK, Ghosh KS, Debnath J, Dasgupta S. Binding of all-trans retinoic acid to human serum albumin: fluorescence, FT-IR and circular dichroism studies. International Journal of Biological Macromolecules. 2006;38(3–5):197–202. PubMed PMID: 16569428
10. Xie M-X, Long M, Liu Y, Qin C, Wang Y-D. Characterization of the interaction between human serum albumin and morin. BiochimicaetBiophysicaActa. 2006;1760(8):1184–1191. PubMed PMID: 16750302
11. Wang Y, Yu H, Shi X, Luo Z, Lin D, Huang M. Structural mechanism of ring opening reaction of glucose by human serum albumin. The Journal of Biological Chemistry. 2013;288:15980–15987 PubMed PMID: 23592780
12. Keshavarz F, Alavianmehr MM, Yousefi R. Molecular interaction of benzalkoniumIbuprofenate and its discrete ingredients with human serum albumin. Physical Chemistry Research. 2013;1(2):111–116.
13. Kanakis CD, Tarantilis PA, Polissiou MG, Diamantoglou S, Tajmir-Riahi HA. Antioxidant flavonoids bind human serum albumin. Journal of Molecular Structure. 2006;798(1-3):69–74.
14. Sudlow G, Birkett D, Wade D. Further Characterization of Specific Drug Binding Sites on Human Serum Albumin. Molecular PharmacologyNovember 1976,12 (6) 1052-1061;
15.Ranjbar S, Shokoohinia Y, Ghobadi S, et al. Studies of the Interaction between Isoimperatorin and Human Serum Albumin by Multispectroscopic Method: Identification of Possible Binding Site of the Compound Using Esterase Activity of the Protein. The Scientific World Journal. 2013;2013:305081. doi:10.1155/2013/305081.
16. Osama K. Abou-Z. and Othman I. K. Characterization of Subdomain IIA Binding Site of Human Serum Albumin in its Native, Unfolded, and Refolded StatesUsing Small Molecular Probes, Department of Chemistry, Sultan QaboosUniVersity, July 2008, 19
17. Mauro F , Stephen C , Enzo T , Monica G , Gabriella F, Pasquale N , Stefania N and Paolo A. Critical Review The Extraordinary Ligand Binding Properties of Human Serum Albumin, IUBMB LifeVolume 57, Issue 12, Version of Record online: 3 JAN 2008
18. Ascenzi P, Masi A, Fanali G, Fasano M. Heme-based catalytic properties of human serum albumin. Cell Death Discovery 1, Article number: 15025, 2015
19. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC et al. UCSF chimera—a visualization system for exploratory research and analysis. J ComputChem 2004; 25: 1605–1612.
20. Meng EC, Pettersen EF, Couch GS, Huang CC, Ferrin TE. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics 2006; 7: 339.
21. Niwa T. Organic acids and the uremic syndrome: protein metabolite hypothesis in the progression of chronic renal failure. SeminNephrol. 1996 May;16(3):167-82. Review. PubMed PMID: 8734460.
22. Tsutsumi Y, Deguchi T, Takano M, Takadate A, Lindup WE, Otagiri M. Renal disposition of a furan dicarboxylic acid and other uremic toxins in the rat. J PharmacolExpTher. 2002 Nov;303(2):880-7. PubMed PMID: 12388676.
23. Niwa T. Recent progress in the analysis of uremic toxins by mass spectrometry.JChromatogr B AnalytTechnol Biomed Life Sci. 2009 Sep 1;877(25):2600-6. doi:10.1016/j.jchromb.2008.11.032. Epub 2008 Nov 27. Review. PubMed PMID; 19083276.
24. Hellerstein MK, Schwarz JM, Neese RA. Regulation of hepatic de novolipogenesis in humans. Annu Rev Nutr. 1996;16:523-57. Review. PubMed PMID: 8839937.
25. Niwa T, Aiuchi T, Nakaya K, Emoto Y, Miyazaki T, Maeda K. Inhibition of mitochondrial respiration by furan carboxylic acid accumulated in uremic serum in its albumin-bound and non-dialyzable form. ClinNephrol 1993; 39: 92–6
26.Deguchi T, Ohtsuki S, Otagiri M, Takanaga H, Asaba H, Mori S, Terasaki T. Major role of organic anion transporter 3 in the transport of indoxyl sulfate in the kidney. Kidney Int. 2002 May;61(5):1760-8. PubMed PMID: 11967025.
27. Niwa T, Yazawa T, Kodama T, Uehara Y, Maeda K, Yamada K. Efficient removal of albumin-bound furancarboxylic acid, an inhibitor of erythropoiesis, by continuous ambulatory peritoneal dialysis. Nephron 1990; 56:241–5.
28. Spiteller M, Spiteller G. Separation and characterization of acidic urine constituents. [author’stransl]. J Chromatogr 1979; 164:253–317.
29. Meert N, Schepers E, De Smet R, Argiles A, Cohen G, Deppisch R, et al. Inconsistency of reported uremic toxin concentrations. Artif Organs 2007;31 :600–11.
30. Mauro F, Stephen C , Enzo T , Monica G , Gabriella F , Pasquale N, Stefania N and Paolo A. Critical Review The Extraordinary Ligand Binding Properties of Human Serum Albumin, IUBMB LifeVolume 57. 2008: 1: Issue 12, Version of Record online:
31. Chimura Y, Takamatsu H, Ideuchi H, Oda M, Takeda K, Saitoh H. [Plasmaconcentrations of anionic uremic toxins in hemodialysis patients and theireffects on protein binding of pravastatin]. YakugakuZasshi. 2015;135(6):821-8. doi: 10.1248/yakushi.14-00244. Chinese. PubMed PMID: 26028417.
32. Sakai T, Takadate A, Otagiri M. Characterization of binding site of uremic toxins on human serum albumin. Biol Pharm Bull. 1995 Dec;18(12):1755-61. PubMed8787801.
33. Carter D.C., Ho J.X. Structure of serum albumin, Adv. Protein Chem., 1994, vol. 45:153-203 PubMed PMID: 8154369
34.Kratochwil NA, Huber W, Müller F, Kansy M, Gerber PR. Predicting plasmaprotein binding of drugs: a new approach. BiochemPharmacol. 2002 Nov1;64(9):1355-74. PubMed PMID: 12392818.
35. Fujiwara S, Amisaki T. Fatty acid binding to serum albumin: molecularsimulation approaches. BiochimBiophysActa. 2013 Dec;1830(12):5427-34. Review. PubMed PMID: 23567799.
36. Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. Structural basis of the drug-binding specificity of human serum albumin. J Mol Biol. 2005 Oct 14;353(1):38-52. PubMed PMID: 16169013.37. Tsutsumi Y, Maruyama T, Takadate A, Goto M, Matsunaga H, Otagiri M.Interaction between two dicarboxylate endogenous substances, bilirubin and an uremic toxin, 3-carboxy-4-methyl-5 propyl-2-
37. Tsutsumi Y, Maruyama T, Takadate A, Goto M, Matsunaga H, Otagiri M.Interaction between two dicarboxylate endogenous substances, bilirubin and an uremic toxin, 3-carboxy-4-methyl-5 propyl-2-furanpropanoic acid, on human serum albumin. Pharm Res. 1999 Jun;16(6):916-23. PubMed PMID: 10397614.
38. Éva A. E, László H, Anasztázia H, Tiziano T, Christian G. H,Bernhard K,Keppler,Tamás K. Interactions of the carrier ligands of antidiabetic metal complexes with human serum albumin: A combined spectroscopic and separation approach with molecular modeling studies. Bioorganic & Medicinal Chemistry 19 (2011) 4202–4210
39. Colmenarejo G, Alvarez-Pedraglio A, Lavandera JL. Cheminformatic models to predict binding affinities to human serum albumin. J Med Chem. 2001 Dec 6;44(25):4370-8. PubMed PMID: 11728183.