This is a basic tutorial on removing the hetero-atoms (HETATMS) and chains from PDB files. It is an essential step for computational and molecular dynamics simulation.
There are two simple ways to remove HETATMS and chains from PDB files.
1. Using a text editor
Removing HETATOMS
- Open your PDB file in an editor such as notepad++ (in Windows) or gedit/notepadqq (in Linux).
- Go to the end of the file. You will see many lines with ‘HETATM’ in the first column from the right (Figure 1).
- Remove these lines. DON’T remove the last two lines (‘MASTER’ & ‘END’).
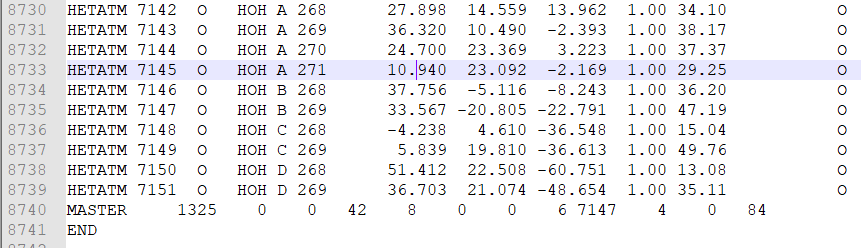
Figure 1 HETATM rows in a PDB file.
Removing Chains
Now, look at the fifth column in Figure 1. As you can see, there are four chains in that protein: A, B, C, and D. Let’s suppose we need chain A only, then we have to remove the rest of the three chains.
- After removing HETATM rows, start removing other chains from the row having ‘TER’ in the first column from the right (Figure 2).
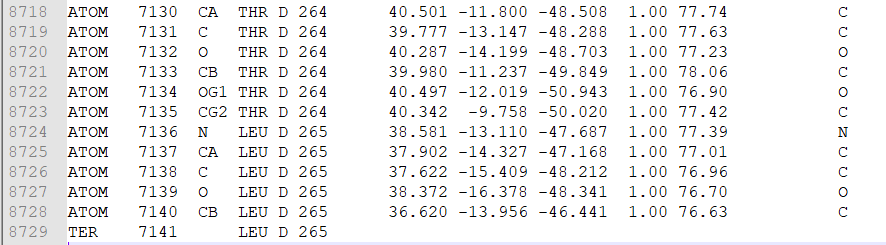
Figure 2 Chain D in the PDB file.
- Keep removing until you reach the line showing ‘TER’ in the first column and ‘A’ in the fifth column (Figure 3). Now, you must be left with the last two rows (MASTER & END) at the end of the file.
- Now, save this file.
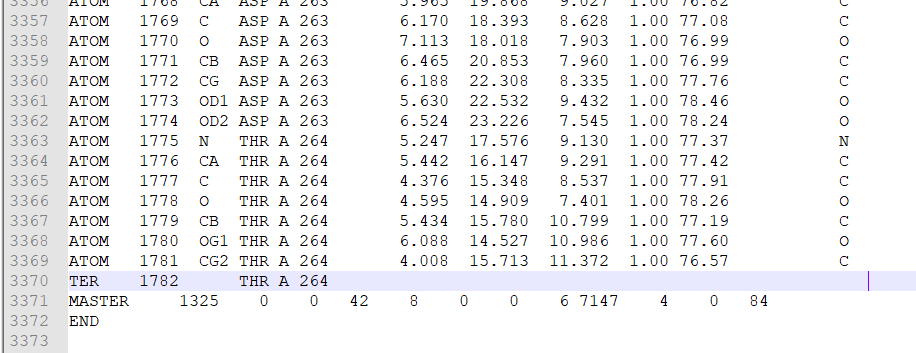
Figure 3 Remaining chain A and the last two rows (MASTER & END) at the end of the file.
2. Using Pymol
The first option seems tedious as compared to using Pymol.
- Open the PDB file in Pymol.
- Go to the bottom left panel. There you will see some options including ‘S’ and ‘F’.
- Click on ‘S’. It will display the chains/ amino acid residues present in that structure.
- Just above these options, you will see: “Selecting: Residues” written there (Figure 4).

Figure 4 Bottom-left panel in Pymol showing the ‘S’ option.
- Left-click with the mouse on Residues one time. It will show ‘Chains’ (Figure 5).
Figure 5 Bottom-left panel in Pymol showing the ‘Chains’ option.
- Let’s suppose you need only chain A. Go to the displayed amino acid residues. Scroll to the end.
- Select the chains that you want to delete by left-clicking with your mouse.
- Go to the top right panel. There you will find three rows including ‘(sele)’. Click on ‘A’ (means action). It will show a small window, select ‘remove atoms’. It will remove the selected chains.
- Now, go to File –> Export Molecule –> PDB Options. Check ‘Write CONECT records for all bonds’ and uncheck ‘Write segment identifier (segi) column’. The latter is optional. Click ‘Save’.
Now, you have successfully removed all HETATMS and irrelevant chains from your PDB file.
Reference
- The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.