We provide a new Python script to visualize a chemical structure using Py3Dmol [1]. You can use this script to visualize protein structures as well. It is explained below.
Search
Have an existing account?
Sign In
How to visualize a 3D structure using Py3Dmol?

AI Tools vs Traditional Tools in Bioinformatics- Which one to select?

We find a lot of bioinformatics tools designed to perform a single task. Their algorithms differ, but they provide relatable results. Recently, we have seen a surge in AI tools in bioinformatics. It becomes challenging to select the most suitable tools for specific tasks, such as sequence alignment and structure prediction. In this article, we will discuss what they are, how they differ, and where each is most useful.
AI vs Physics in Molecular Docking: Towards Faster and More Accurate Pose Prediction

Abstract
Molecular docking is a computational technique used in drug discovery to predict how small molecules (ligands) fit into the three-dimensional binding site of a target protein. By modeling these interactions, researchers can rapidly prioritize compounds that are most likely to bind strongly to the target, accelerating the design of new therapeutics. Physics-based docking relies on empirical force fields and can take minutes per compound. By contrast, AI-driven docking can predict a final ligand pose in a few seconds and inherently captures complex interaction patterns. AI delivers a higher fraction of correct poses. Even greater gains emerge when AI’s predicted poses are followed by a brief physics-based minimization step (exemplified with ArtiDock+UFF and ArtiDock+Vina in the article), raising overall accuracy further without adding substantial computation time. In challenging cases, such as binding sites with metal ions, organic cofactors, or structured water molecules, this AI+physics combination demonstrates markedly better accuracy and interaction fidelity.
Introduction
Molecular docking is a computational technique used in drug discovery to predict how small molecules (ligands) fit into the three-dimensional binding site of a target protein, a biomolecule whose malfunction or dysregulation causes a disease [1]. By modeling these interactions, researchers can rapidly prioritize compounds that are most likely to bind strongly to the target, accelerating the design of new therapeutics.
Physics-based docking methods rely on established physical principles: modeling electrostatics, van der Waals forces, hydrogen bonds, and desolvation via empirical force fields. These algorithms systematically explore ligand placements and compute an energy score for each. While they have powered virtual screening for decades, they can take minutes per compound and often struggle with challenging cases like metal-coordinating sites or structured water networks [2]. By contrast, AI-driven docking replaces hand-tuned energy functions with neural networks trained on thousands of known protein-ligand complexes. A trained AI model can predict a final ligand pose in a few seconds, enabling the rapid screening of millions of compounds, and inherently captures complex interaction patterns that are hard to encode in traditional force fields [3].
Overall Outperformance
To assess the performance of protein-ligand docking methods, researchers rely on benchmarks, curated collections of experimentally solved complexes that span diverse protein families and ligand chemistries. By running each algorithm on the same set and comparing predicted ligand poses against the true binding modes, we obtain directly comparable accuracy. In our study, we utilized the PoseX benchmark dataset, which reflects the variety of challenges encountered in real-world drug-discovery
projects. Results are presented in Figure 1.
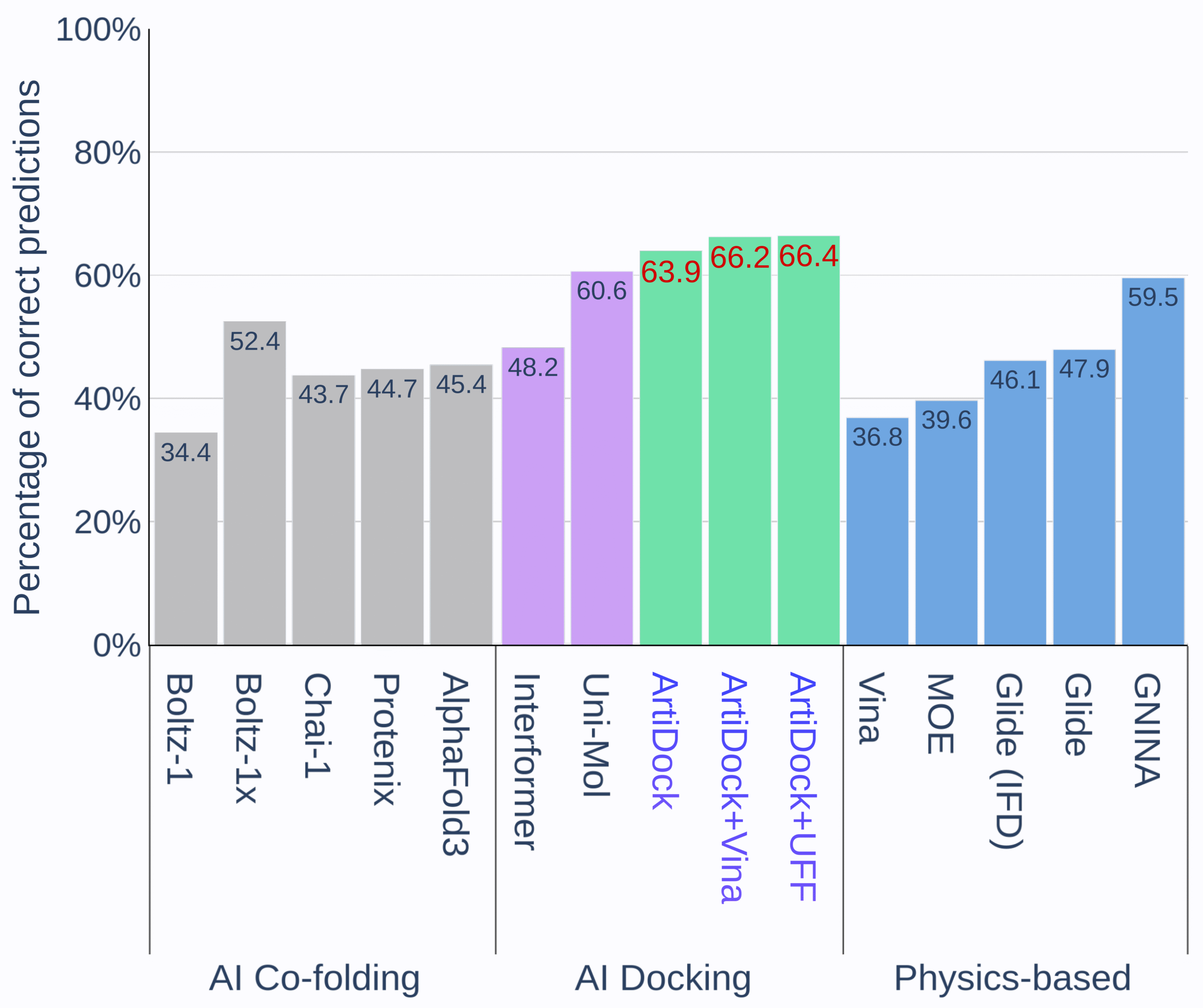
Figure 1. Percentage of correct predictions by protein-ligand docking methods in PoseX benchmark. [Credit: Receptor.AI]
Pure AI-driven docking engines such as ArtiDock and Uni-Mol deliver a higher fraction of correct poses than classic force-field methods like AutoDock Vina or Glide. By learning directly from thousands of solved structures, these AI models capture intricate interaction patterns that rigid scoring functions often miss, translating into consistently superior pose-prediction rates. Even greater gains emerge when ArtiDock’s AI-predicted poses are followed by a brief physics-based refinement. In this hybrid workflow, ArtiDock first proposes the most likely binding geometry within seconds, and then either Vina or Universal Force Field (UFF) minimization smooths out any remaining steric clashes or strain. Because the neural network has already pinpointed the correct region and orientation, this additional step raises overall accuracy further without adding substantial computation time (ArtiDock+Vina and ArtiDock+UFF on Figure 1).
Co-folding models like AlphaFold 3, Boltz-1x, and Protenix aim to predict the protein pocket and dock the ligand in one go. While this unified approach can be useful, especially for targets without high-resolution structures, their current accuracy remains below both specialized AI docking and physics-based methods [4].
Performance in Difficult Areas
Docking becomes especially challenging when the binding site contains non-protein entities such as metal ions, organic cofactors, or structured water molecules. In these scenarios, traditional force-field methods struggle because generic parameter sets often fail to capture the precise coordination geometry of metal centers, the directional hydrogen-bonding networks of water bridges, and the unique electronic environments around cofactors. As a result, physics-based tools like AutoDock Vina or Glide can misplace ligands or generate poses that clash sterically or lack key interactions, limiting their reliability in realistic drug discovery pockets [2, 5, 6].
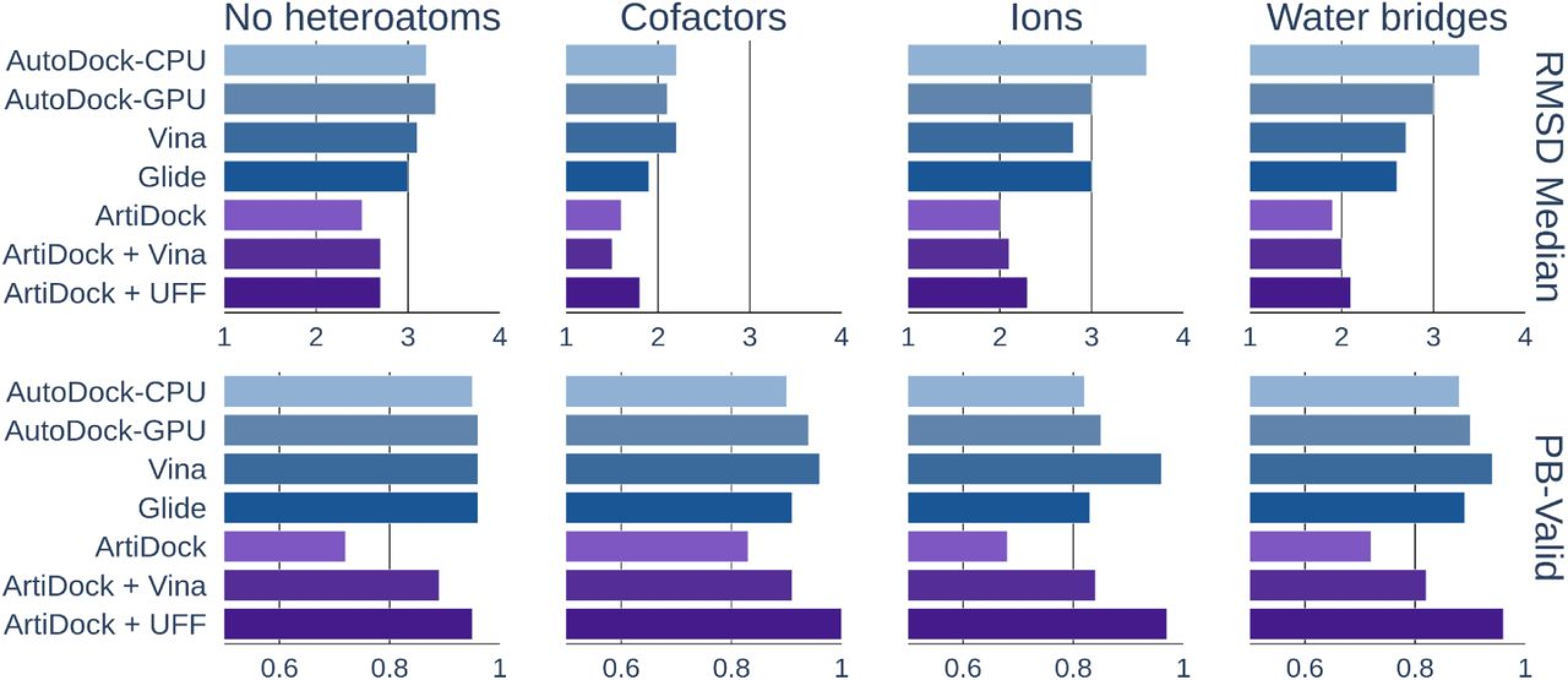
Figure 2. ArtiDock performance in pockets containing no heteroatoms, cofactors, ions, or bound water: median RMSD (root-mean-square deviation, in Å) and PB-Valid (fraction of poses passing PoseBusters quality checks). [Credit: Receptor.AI]
In contrast, ArtiDock demonstrates markedly better accuracy and interaction fidelity across all tested difficult-site categories (Figure 2). By learning directly from diverse structural examples, including those with metals, water networks, and cofactors, its neural network internalizes the complex patterns of coordination and solvation that traditional scoring functions miss. Consequently, ArtiDock yields lower median RMSD values and reproduces more of the true protein-ligand contact network, maintaining correct geometry even before any additional refinement.
Adding a short physics-based minimization step using either Vina rescoring or Universal Force Field (UFF) energy minimization introduces a small uptick in RMSD, since the energy optimizer may subtly shift the pose. However, this trade-off is counterbalanced by a significant increase in chemical validity, restoring or strengthening key interactions.
How It Works: ArtiDock Architecture
Receptor.AI’s ArtiDock starts by converting the protein binding site and the small molecule ligand into simple graph representations (workflow in Figure 3). The ligand is modeled as a network of atoms connected by bonds, while the protein pocket is captured as a set of nearby heavy atoms annotated with basic chemical and spatial features: enough to describe the pocket’s shape and environment without overwhelming detail [7].
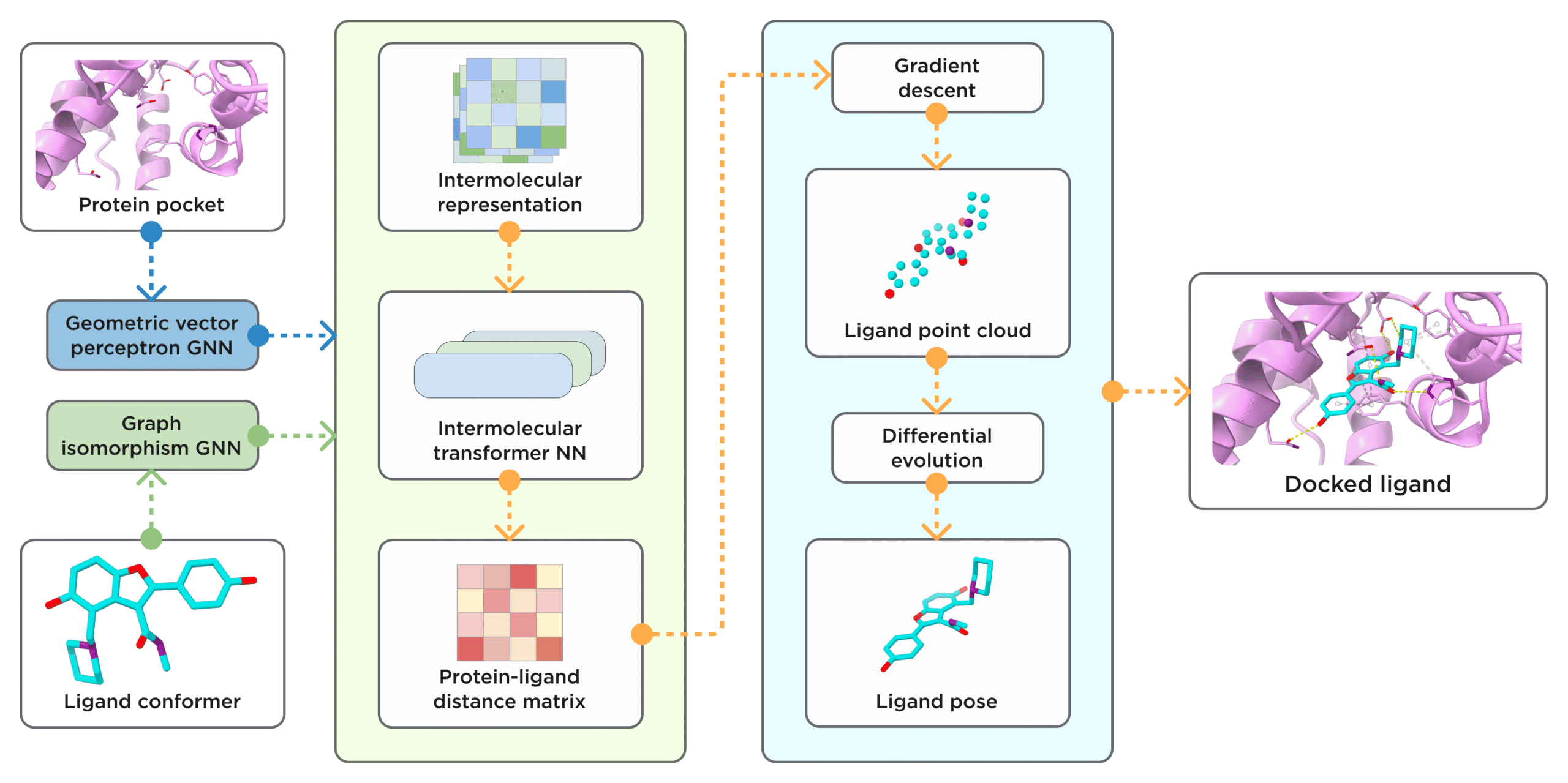
Figure 3. General scheme of ArtiDock model architecture and inference pipeline. [Credit: Receptor.AI]
A lightweight neural network then learns how these two graphs fit together. By examining many examples of known protein–ligand complexes, ArtiDock infers the ideal distances between each ligand atom and the surrounding pocket atoms. This learned distance matrix effectively encodes the best way for the ligand to nestle into the target site [7].
Finally, a fast procedure converts the predicted distances into a three-dimensional pose. It aligns a pre-generated ligand conformation to match the learned distance pattern, producing a realistic placement of the compound in the pocket. This direct prediction approach bypasses the lengthy trial-and-error cycles of traditional docking, delivering accurate poses in a fraction of the time [7].
Conclusion
AI-driven docking tools like ArtiDock have shown that models trained on large, diverse structural datasets can overcome the speed and accuracy limits of traditional physics-based methods. By delivering precise poses in seconds, even in the most challenging binding sites, these AI approaches promise to accelerate drug discovery.
Further details regarding ArtiDock can be found here.
References
1. Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Reviews Drug Discovery. 2004;3(11):935–949. doi:10.1038/nrd1549
2. Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: New docking methods,expanded force field, and Python bindings. Journal of Chemical Information and Modeling. 2021;61(8):3891–3898. doi: 10.1021/acs.jcim.1c00203
3. Alcaide E, Gao Z, Ke G, et al. Uni-Mol Docking V2: Towards realistic and accurate binding pose prediction. arXiv. 2024;2405.11769. doi: 10.48550/arXiv.2405.11769
4. Abramson J, Adler J, Dunger J et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024). https://doi.org/10.1038/s41586-024-07487-w
5. Riccardi L, Genna V, De Vivo M. Metal–ligand interactions in drug design. Nat Rev Chem. 2018;2(7):100–112. doi: 10.1038/s41570-018-0018-6
6. Hu X, Maffucci I, Contini A. Advances in the Treatment of Explicit Water Molecules in Docking and Binding Free Energy Calculations. Curr Med Chem. 2019;26(42):7598-7622. doi:
10.2174/0929867325666180514110824
7. Voitsitskyi, T., Koleiev, I., Stratiichuk, R., Kot, O., Kyrylenko, R., Savchenko, I., Husak, V., Yesylevskyy, S., Starosyla, S., & Nafiiev, A. (2025). ArtiDock: accurate machine learning approach to protein–ligand docking optimized for high‑throughput virtual screening. bioRxiv, 2024.03.14.585019 (v2). https://doi.org/10.1101/2024.03.14.585019
10 years of Bioinformatics Review: From a Blog to a Bioinformatics Knowledge Hub!

We are celebrating 10 years of Bioinformatics Review! It has been a decade since this platform came into existence. Every platform has a story. Bioinformatics Review is not just a blog, but a journey I’ve been fortunate to witness closely. Today, we are proud to share the journey of Bioinformatics Review- from a blog to a true Bioinformatics Knowledge Hub.
Birth of Bioinformatics Review
It all started in early 2015 when we had an idea of creating a platform of knowledge for bioinformatics, as no such platforms existed a decade ago. His vision was clear:
to create an independent platform where researchers and students could access knowledge, tutorials, and updates in bioinformatics.
For many students starting out, bioinformatics felt confusing. Since it is an interdisciplinary field, it was challenging for the students who were solely coming from either biology or computer science. Therefore, we came up with the idea of creating this platform.
We started with a very few articles, trying to cover the then-current topics. At that time, it took us hours to read and understand the research articles (being young researchers) to ensure the clarity and gist of the latest research in the form of an article. Late nights, continuous research, and endless brainstorming about how to make bioinformatics easier for others. Now it has hundreds of bioinformatics articles that are most appreciated by our readers.
How has Bioinformatics Review helped me shape my career?
Bioinformatics Review has always helped me grow by learning new skills and tools. The articles and tutorials that I have contributed to have sharpened my understanding of bioinformatics concepts and improved my scientific communication skills. I have collaborated with professors and researchers from elite universities. I have shared projects with them that helped me to gain experience in different domains of bioinformatics. Bioinformatics Review is a great medium to streamline my computational work, which can reach more people struggling through the same issues in their research.
Being a part of this platform not only enhanced my bioinformatics skills but also shaped my approach to research, collaboration, and mentorship. It has also helped me build a professional network in the bioinformatics community. For anyone starting in bioinformatics or trying to make their way through bioinformatics, having a resource like Bioinformatics Review can be truly transformative.
Journey Ahead
It started as a small personal initiative, which later grew into something bigger. It has become a space where researchers can find answers, learn bioinformatics, and apply it in their scientific journey. Over the years, Bioinformatics Review has reached readers across the world, covering diverse topics: from step-by-step tutorials, reviews, research articles, and tips and tricks to accompany you in your research career. Many researchers have told me how this platform has helped them bridge the gap between theory and practice in their work. Your appreciative emails really keep us going.
We’ve also expanded beyond tutorials and articles. We have successfully conducted internship programs and collaborative research. Unfortunately, our first group project couldn’t get published due to the lack of funding. We hope to get funding for our future projects. Our Python packages are available on GitHub to help researchers implement bioinformatics more easily, making tools and scripts accessible to everyone.
We have received several suggestions and appreciation from our readers all over the world, including some interesting topics to write more articles about. We are currently working on the suggested topics and will soon make them accessible to all. If you still have any specific topic you would like to read about, write us at info@bioinformaticsreview.com. Besides, BiR would like to welcome new authors who are interested in bioinformatics and who share their knowledge worldwide.
We are glad that today, Bioinformatics Review stands not just as a blog but as a bioinformatics knowledge hub. We will be focusing on Machine Learning and AI in bioinformatics this year. You will get to read and watch more interesting and useful bioinformatics content. And this is only the beginning. We have many more ideas in store for the future. You are going to enjoy bioinformatics even more! Stay tuned for exciting updates ahead!
Starting in Bioinformatics? Do This First!

I receive a lot of DMs asking for career guidance in the field of bioinformatics. This brief article addresses frequently asked questions about a career in bioinformatics.
Bioinformatics can be particularly challenging for beginners seeking to establish a career in this field. While I truly appreciate your interest in the field, I highly recommend doing some initial research before reaching out. I expect a bit of beginning knowledge from those who are sending emails and DMs. This can be obtained by simply Googling the basic concepts. This is even necessary when you are trying to start a career in a field. You have to have a basic knowledge of the field. Therefore, before directly asking questions, please try searching online.
A good starting point could be:
- Reading beginner-friendly articles on bioinformatics concepts
- Watching free lectures or YouTube tutorials
- Taking short online courses to grasp foundational skills
- Joining relevant forums or LinkedIn groups to observe ongoing discussions
I have written a few articles previously to address this issue. Here I am linking some articles that will help you kickstart your journey into bioinformatics.
- https://bioinformaticsreview.com/20210517/basic-bioinformatics-concepts-to-learn-for-beginners/
- https://bioinformaticsreview.com/20200825/list-of-bioinformatics-books-for-beginners/
- https://bioinformaticsreview.com/20201002/careers-in-bioinformatics-and-computational-biology/
- https://bioinformaticsreview.com/20200922/current-research-topics-in-bioinformatics/
- https://bioinformaticsreview.com/20200818/bioinformatics-where-how-to-start/
- https://bioinformaticsreview.com/20201207/bioinformatics-is-it-a-magical-research-field/
This basic groundwork will not only help you ask more specific and meaningful questions but also show that you are genuinely interested and willing to put in the effort.
Secondly, I strongly suggest taking some online courses on bioinformatics topics to enhance your skills and strengthen your resume. This will demonstrate that you have a solid foundational knowledge of the subject, which can help with professional recognition.
Bioinformatics is a vast and exciting domain, and building a foundation through self-learning, such as reading articles, watching tutorials, and exploring beginner courses, will make your career journey smoother and more productive. To summarize, do your homework first, then seek advice. This combination will give you the best results in shaping your career in bioinformatics.
[Editorial] Is it ethical to change the order of authors’ names in a manuscript?

A manuscript is not just a collection of paragraphs, it is a result of hard work by researchers, particularly the first author. As reflected in its content, it can be a work of a few months to a few years. This work is what makes a manuscript worthy of publication in reputed journals. For an author, especially the first author, a manuscript can mean the world, which can eventually lead to the graduation of a Ph.D. scholar or may serve as a ticket to admission into a prestigious university for a Master’s student.
[Tutorial] Installing BBTools on Ubuntu (Linux).

BBTools is a suite consisting of various bioinformatics tools for DNA and RNA sequencing data analysis [1]. It can process various file formats such as fasta, fastq, sam, compressed, or raw. This suite is written in JAVA and works on Windows, Linux, and MacOS. In this article, we will install it on Ubuntu (Linux).
wes_data_analysis: Whole Exome Sequencing (WES) Data visualization Toolkit

Whole Exome Sequencing (WES) is a genomic technique that sequences only the protein-coding regions (exons) of the genome. In this article, we introduce a new Python package that allows the easy visualization of WES data.
[Editorial] Prioritizing Quality Over Quantity: Safeguarding the Integrity of Academic Research

In recent years, the academic and research communities have witnessed an unprecedented surge in the number of publications. While this growth may initially appear to signify a flourishing intellectual landscape, a deeper examination reveals a troubling trend: the proliferation of publications often comes at the expense of quality. This editorial explores the factors driving this phenomenon, its negative impact on the research community, and the need for systemic reforms to uphold the integrity of scientific research.
[Tutorial] Installing Exabayes on Ubuntu (Linux).

ExaBayes is a software tool designed for large-scale Bayesian phylogenetic inference [1]. It is particularly optimized for analyzing massive datasets, such as those derived from next-generation sequencing, by efficiently estimating phylogenetic trees. ExaBayes employs parallel computing techniques to handle computationally intensive tasks, making it suitable for high-performance computing environments. In this article, we will install Exabayes on Ubuntu (Linux).
How to see ligand interactions and label residues in DS Visualizer?

Discovery Studio (DS) Visualizer [1] is a powerful molecular modeling tool widely used in computational chemistry and bioinformatics. Developed by BIOVIA, it provides an intuitive graphical interface for visualizing and analyzing molecular structures, including docking output analysis. In this article, we provide step-by-step instructions for viewing ligand interactions and labeling interacting residues in DS Visualizer. Additionally, you can explore several other articles on docking analysis and using DS Visualizer.
[Tutorial] How to install openbabel on Ubuntu (Linux)?

Video Tutorial: Fix broken Vina output in DS Visualizer.

This is a video tutorial on fixing the Vina docking output in DS Visualizer.
How to Fix Broken Vina Docked Output in DS Visualizer: A Step-by-Step Guide

Molecular docking is a crucial method in computational drug discovery, allowing researchers to predict the binding of small molecules (ligands) to their target proteins. AutoDock Vina is a popular tool for this purpose, producing docked poses as output files. However, when these output files are loaded into visualization tools like DS Visualizer (Discovery Studio Visualizer), users sometimes encounter issues such as broken structures or scattered fragments. In this article, we provide a new Python script to solve this issue.
How to install & execute Discovery Studio Visualizer on Ubuntu (Linux)?

DS Visualizer is a comprehensive, free molecular modeling and visualization tool designed by BIOVIA, part of Dassault Systèmes [1]. It enables researchers to visualize and analyze complex chemical and biological data, including molecular structures, sequences, and simulations.DS Visualizer’s user-friendly interface supports various file formats and provides powerful tools for molecular editing, docking, and structure analysis. In this article, we are installing DS Visualizer on Ubuntu (Linux).
[Tutorial] Installing HTSlib on Ubuntu (Linux).
![[Tutorial] Installing HTSlib on Ubuntu (Linux).](https://bioinformaticsreview.com/wp-content/uploads/2024/08/htslib.jpg)
HTSlib is an open-source C library designed for handling high-throughput sequencing (HTS) data [1]. It provides the underlying functionality for manipulating various file formats commonly used in genomics, such as SAM (Sequence Alignment/Map), BAM (Binary Alignment/Map), CRAM (Compressed Reference-oriented Alignment Map), and VCF (Variant Call Format). In this article, we are installing on Ubuntu (Linux).
List of widely used MD Simulation Analysis Tools.

Molecular Dynamics (MD) simulation analysis involves interpreting the vast amounts of data generated during the simulation of molecular systems. These analyses are necessary to study the physical movements of atoms and molecules, the stability of molecular conformations, reaction mechanisms, and thermodynamic properties, among other aspects. In this article, we will give a brief overview of some widely used MD simulation analysis tools.
[Tutorial] Installing ProteStAr on Ubuntu (Linux).

ProteStAr is a bioinformatics tool to compress protein structure files [1]. It compresses PDB/CIF files and supplementary PAE files. The compression is lossless. However, users are allowed to generate the lossy compression of files. In this article, we are installing ProteStar on Ubuntu.
DockingAnalyzer.py: A New Python script to Identify Ligand Binding in Protein Pockets.

High-throughput virtual screening (HTVS) is a pivotal technique in drug discovery that screens extensive libraries of compounds to find potential drug candidates. One of the essential tasks in HTVS is to ensure that ligands are binding within the protein’s binding pocket. This task can be particularly challenging when dealing with thousands of docking results. To address this challenge, we present a Python script that automates the analysis of molecular docking results generated by AutoDock Vina [1] using PyMOL [2]. This script calculates the center of mass (COM) for each docked pose, compares it with a reference ligand’s COM, and identifies poses that bind within a specified threshold distance. This process is crucial in mass docking scenarios where confirming ligand binding within the pocket is necessary. (more…)
How to copy and rename files simultaneously in same directory in Ubuntu (Linux)?

Copying and renaming files from one destination to another is easily accomplished by the cp command in Linux. However, it becomes more complex when we want to copy and rename all files in the same directory while assigning a serial number to each copy. In this article, we provide a shell script to copy and rename files present in a directory, adding a unique serial number to each copy.