As the needs say the importance of sequencing of genomes, it is equally important to visualize them. There exists some tools to visualize the genomes,but they are static and standalone, and very much complex to install and use. Newer tools are required to ease the visualization of genomes utilizing various new features and which are more interactive. GenomeD3 Plot is a newly created visualization library written in Java script. It uses the D3,i.e., Data Driven Documents Library which is used to produce dynamic, interactive data visualizations in web browsers. GenomeD3Plot is very user-friendly and allows to interact with data, dynamical view alteration is possible,and easy resize or reposition the visualization in the browser.
The goal of R Laird Matthew was to create a library with minimal external dependencies that could be integrated in to existing web applications just as a developer might include an image or table.
GenomeD3 Plot uses the JSON configuration which is a standardized and well supported data format, that reduces the complexity of use and provide better visualization. The image will be created in SVG format and can be easily imported to PNG format as per the requirements.
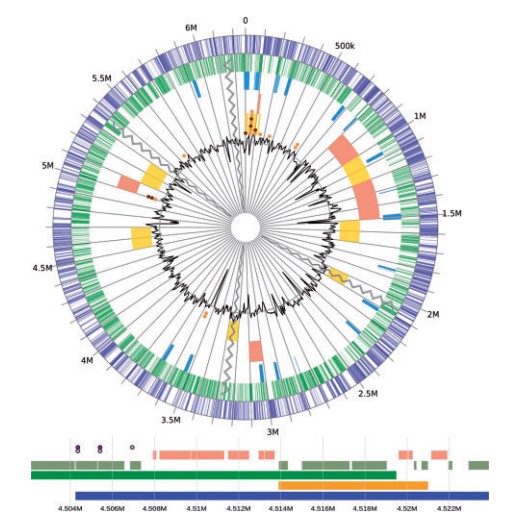
Fig.1 GenomeD3Plot circular and linear visualization of an example genome
with annotation data
With GenomeD3 Plot, the genome can be viewed in differnt tracks, such as if one wish to view a specific base pair or a series of base pairs to visualize GC content, or more.GenomeD3 Plot provides a rich API ( application program interface that specifies how software components should interact) to dynamically manipulate visualization.
A linear and circular plot can also be tied together so that manipulation of one will cause a mirror alteration in the other, such as zooming or changing the visible region of the genome. A specific region can be recentre to focus. Many other features have been introduced in GenomeD3 Plot for easy visualization and interpretation of genomes.
Note:
An exhaustive list of references for this article is available with the author and is available on personal request, for more details write to muniba@bioinformaticsreview.com.