Secondary structure formation and conformational changes play a key role in understanding molecular evolution and its functional aspects. Among DNA, RNA, and Protein, secondary structure analyses of RNA and Proteins have attracted a lot of research and development.
RNA secondary structure has been proved necessary to understand the regulatory functions of microRNAs, and to infer or understand the function of an RNA molecule, understanding the ability of small RNAs to regulate gene expression and so on. It is leading to an increasing demand for structured RNAs in the genomic and transcriptomic context which is a difficult task. In this case, RNA local pairwise alignment may be helpful for which some of the tools are available.
Foldalign is one such tool which implements the local pairwise RNA structural alignment based on the Sankoff algorithm (Sankoff, 1985). CMfinder also performs the pairwise alignment but it is not local. Sankoff algorithm is complex and cannot handle long sequences. Foldalign overcomes these difficulties. It can align long sequences, less complex and requires less processing time and memory.
Sundfeld et al., (2015) has developed a multithreaded parallel algorithm which has been implemented in C++.
ALGORITHM:
Foldalign 2.5 version is a multi-threaded implementation of local pairwise structural alignment of RNA. It uses only six nested loops and calculates the alignment in parallel for many pairs. The dynamic programming matrix is divided into two memories: Long-term memory and Short term memory. Long-term memory consists of the cells that can only be a part of a multi-branch loop and short-term memory consists of the other cells. In this multithreaded version of Foldalign, various threads of particular lengths are created and each thread works on its own value of the cell. It starts from the length of the sequence up to the last single cell, i.e., i=L1, L1-1,…..1 (for one sequence, named S1) and k= L2,L2-1,…..1 (for another sequence, S2). Many cells are calculated in parallel. Every thread calculates each and every cell sequentially starting from first thread of one sequence to its full length and then restarts from the first thread of the second sequence, such that it aligns cell to cell or in other words, aligns residue to residue implementing local pairwise alignment.
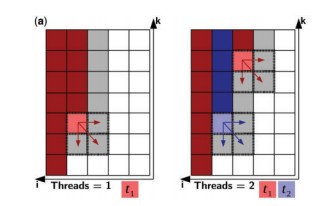
Fig.1 (a) Parallel design example of two sequences. Every cell corresponds to a bidimensional matrix. Red and blue are cells processed by threads t1 and t2, respectively. Dark red/blue are cells that have already been processed, light red/blue are cells being processed and white or grey are cells to be processed next. The dashed area represents cells that are being read and written by one thread.
Foldalign can take larger sequences as input and completes the structural alignment in a reasonable time and saves the computer memory, and it is able to produce accurate predictions.
For further reading, click here.
References:
Foldalign 2.5: multithreaded implementation for pairwise structural RNA alignment.
Sundfeld D1, Havgaard JH2, de Melo AC3, Gorodkin J2
NOTE:
For any query, please write to muniba@bioinformaticsreview.com