In this rising era of personalized medicine, drug discovery, data refining, chemical compound databases, drug information, identification of symptoms by various levels of biomarkers, bioinformatics have been playing a pivotal role in bringing out the best deliverables in a cost and time effective manner. There are various chemical databases available to provide information about various drugs, their pharmacokinetics, pharmacodynamics, drug interactions, etc., which in turn become the major resource for a computational approach to drug discovery. Some examples of such data repositories are PubChem [1,2] (a database composed of PubChem Substance, PubChem Compound and PubChem Bioassay), DrugBank [3], ChEBI [4] , SIDER [5](Side Effect Resource), ChemSpider [6], ChemExper [7], TTD [8] (Therapeutic Drug Target Database).
DrugQuest [9] is a web-application which applies the knowledge discovery and text mining techniques to parse the DrugBank repository and group drugs according to the textual information.
DrugQuest is a text mining tool which extracts the useful information from the available data. It specifically extracts data from the DrugBank only by clustering all the records according to the textual information provided such as symptoms, diseases, pathways, toxicity, etc. [9] (Fig.1).
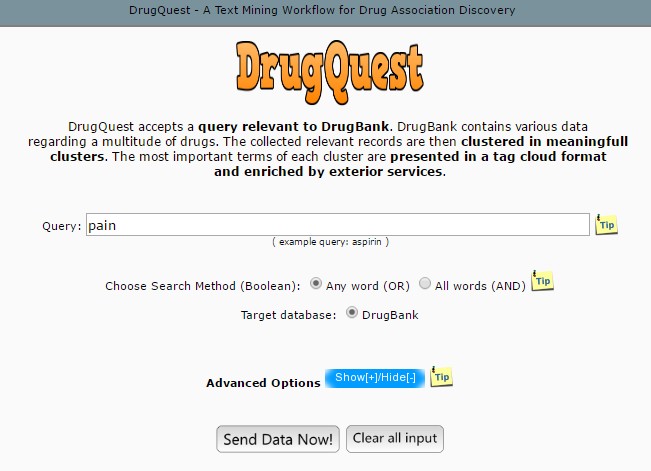
Fig.1 Homepage of DrugQuest. [http://bioinformatics.med.uoc.gr/cgi-bin/drugquest/drugQuest.cgi]
Workflow of DrugQuest:
1. A query is entered by the user which is searched in Boolean operations, i.e., all words (AND) and any word (OR). The query may be any word related to the drug or disease to be searched such as the symptoms, headache, pain, aspirin, etc.
2. DrugBank records are searched based upon the query.
3. Textual entries present in DrugBank are matched with the query and drug records are retrieved.
4. Applies TextQuest algorithm to identify non-tagged Significant Terms.
5. TF-IDF score (product of Term Frequency and Inverse Document Frequency) is calculated for each non-tagged terms in the records to estimate its importance (i.e., whether it should be included in the results or not).
6. English words with low TF-IDF value are removed on the basis of British National Corpus (BNC -http://www.natcorp.ox.ac.uk/)
7. All the common English words such as articles, prepositions, etc are removed and now the remaining words after the steps 4-7 are considered as “Significant terms”.
8. Each DrugBank record is represented in the binary vector form, i.e., in the form of binary codes, 0 and 1, where 0 defines the absence of significant terms and tagged terms, and 1 defines the presence as shown in Fig.2.
9. DrugBank records are clustered using various clustering algorithms.
10. The results are represented and visualized in two forms: “Tag Clouds” and “Clustered Drugs”.

Fig. 2 Workflow of DrugQuest [10]
TextQuest algorithm is used to identify the significant words, for which it first calculates the TF-IDF score for each word in the database to determine its significance, then removes the words with low TF-IDF score and the common English words [10].
Tag Clouds view displays the result in the form of a cloud highlighting the Significant terms. The font size is proportional to the frequency of the query terms present in the records. The Clustered Drugs form organizes the DrugBank records in different categories with a link to respective DrugBank record. DrugQuest has a limit of 5000 textual records per analysis [10].
References:
1. Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37(Web Server issue):W623–33.
2. Li Q, Cheng T, Wang Y, Bryant SH. PubChem as a public resource for drug discovery. Drug Discov Today. 2010;15(23–24):1052–7
3. Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006 Jan 1;34(Database issue):D668-72. 16381955
4. Degtyarenko K, de Matos P, Ennis M, Hastings J, Zbinden M, McNaught A, Alcantara R, Darsow M, Guedj M, Ashburner M. ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008;36(Database issue):D344–50.
5. Kuhn M, Campillos M, Letunic I, Jensen LJ, Bork P. A side effect resource to capture phenotypic effects of drugs. Mol Syst Biol. 2010;6:343
6. http://www.chemspider.com (accessed on date 17th June 2016)
7. www.chemexper.com (accessed on 17th June 2016)
8. Chen X, Ji ZL, Chen YZ. TTD: therapeutic target database. Nucleic Acids Res. 2002;30(1):412–5.
9. http://bioinformatics.med.uoc.gr/cgi-bin/drugquest/drugQuest.cgi
10. Papanikolaou et al. BMC Bioinformatics 2016, 17(Suppl 5):182