Virtual High Throughput Screening (vHTS), also known as Virtual Screening (VS) is one of the essential steps involved in in-silico drug designing. There are several bioinformatics tools that facilitate the virtual screening of thousands of compounds such as GOLD, GLIDE, Autodock Vina, and so on.
Despite all the processing done by these tools, a basic methodology is required for vHTS of thousands of compounds present in a database. In this article, we are presenting a few basic steps required for performing structure-based VS using bioinformatics tools. The complete schema is presented in Figure 1.
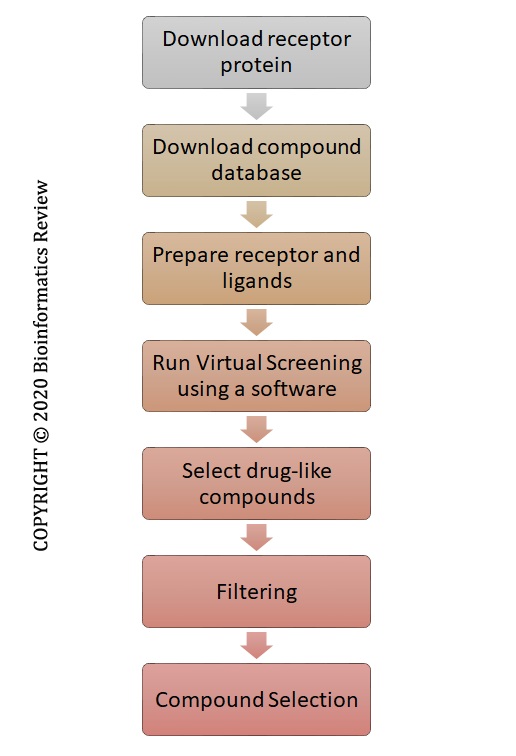
Figure 1 Basic steps involved in virtual screening.
Step 1. Prepare receptor
Download the structure of receptor protein from PDB. In case, the structure is not available, predict the structure using homology modeling or ab-initio methods of structure prediction. Search the literature to gather information about the binding pocket or binding residues of the protein of interest. Select a chain (or two) consisting of the binding region. If the structural details are not available in the literature, then go for binding pocket prediction using webservers or tools.
Step 2. Download compound databases such as the ZINC database of small molecules.
Download the .sdf or .mol2 files of compounds. Some databases allow downloading only smiles of the compounds, then download the smiles and use a web server or software to convert these smiles into .sfd/.mol2 files.
Step 3. Prepare receptor and ligands for docking
Depending upon the software you are using for VS, prepare the files of the receptor protein and the ligands. for example, Autodock Vina requires receptor and ligand files in .pdbqt format.
Step 4. Run VS
Load all the files in the software and run VS. Wait for the results.
Step 5. Select drug-like compounds
Select the drug-like molecules based on the binding affinity and interaction with binding residues. You can also select some lead-like molecules showing less binding affinity.
Step 6. Filtering
You can further filter obtained drug-like molecules based on their toxicity, ADME properties, poses, and so on.
Step 7. Compound selection
Select potential drug candidates for further in vitro experiments.
These are the basic steps involved in VS for structure-based drug designing. There are several bioinformatics tools other than those mentioned above that are used in VS including Discovery Studio, LigandScout, and so on.