The word “Homology modeling”, means comparative modeling or sometimes it is known as Template-Based Modeling (TBM), which refers to develop a three dimensional model of a protein structure by extracting the keen information’s from already experimentally known structure of a homologous protein (the template). The 3D Protein structural information provides great assistance to study the function of different proteins, ligands interactions, dynamics, and Protein-Protein Interactions (PPI). The structure of proteins developed by homology modeling comprises significant information of the 3D spatial arrangement of important amino acid residues in the protein, in this way, it helps to gain insightful knowledge to design new experiments. In the pharmaceutical industry, homology modeling has paramount importance specifically in structure-based drug designing and discovery process. Protein structural elucidation through crystallography experiment is a challenging task in terms of “fine crystals development” and “low resolution” structure development. In this context, protein structure prediction methods have invited much attention. Therefore, in all these available methods, in-silico based homology modeling usually predicts the fastest, efficient and reliable results, which is based on the observation that two proteins associated with the same family (having a similar sequence of amino acids), will possess similar 3D structures. Following are the steps required for homology modeling.
- Template identification
- Target and template amino acid sequence alignment
- Alignment correction
- Backbone making
- Generation of loops
- Side chain generation & optimization
- Ab initio loop building
- Overall model optimization
- Model verification, Quality criteria, model quality
After searching for the most suitable and identical template of protein, the next step is to perform multiple sequence alignment, which includes target and template sequence alignment. It is also equally important to predict the secondary structure. Specifically, the positions of insertions and deletions of amino acid need to be correct (secondary structure, outside regions), likewise, the conserved residues, for instance, active site amino acid must be aligning over each other. When the sequence analysis is completed and the alignment is finalized accordingly, then we may proceed to the final step of model building. Software usually possesses its own program for sequence alignment. The sequential steps of the modeling software may include, generation of backbone, the building of missing parts (e.g. loops), side chains generation for residues, side chain conformational optimization, and finally energy minimization of the model. In our design study, we used SWISS-MODEL WORKSPACE software, which works efficiently with ProMod3 (a comparative modeling engine) [1-2]. This is relatively fast and allocates a nice model quality assessment. The greater the sequence similarity exists between the model and the template the better will be the predicted quality of the model. The server also generates the assessment of model quality output reports.
Material & Methods
The software Swiss Modeler Workspace was used for Homology modeling, which is the free online available tool and can be easily accessible through the link, https://swissmodel.expasy.org/.
Results & Discussion
Homology modeling through SWISS-MODEL WORKSPACE Server
Swiss model workspace server is accessible through online registration or can be directly available for using its tools. Following is the step by step description for homology modeling.
Protein-ligand interactions
Protein-ligand interactions can be demonstrated through, seven types of possible non-covalent existing interactions which may include, [1] hydrogen bonding, [2] hydrophobic contacts, [3] π-π stacking interactions, [4] cation-π interactions, [5] presence of salt bridges, [6] presence of water bridges, [7] presence of halogen bonds [3].
The target protein sequence can be either fetched directly through the PDB code (3A4A), or it can be directly uploaded or paste the FASTA sequence of the target protein on Swiss modeler as follows (Fig. 1).
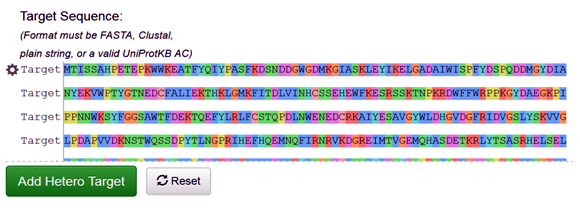
Fig. 1 Depicting the FASTA sequence of the target protein (3A4A) on the SWISS-MODEL
The SWISS-MODEL Template Library (SMTL) Search
The SWISS-MODEL template library consists of experimentally known protein structural database derived from the Protein Data Bank (4-6). It maintains the main repository of structural information for the modeling by providing sequence similarity search to identify and profile the databases through BLAST and HHblits programs [7-8]. The SWISS-MODEL server provides quick access for searching the templates, in parallel mechanism with HHblits and BLAST programs, to search and identify the most suitable template for target-template alignments. The common strategy using these two approaches assures good alignments at higher and lower sequence identity levels, when we click on search for templates button, it will ultimately direct and search the template proteins in Swiss Model Template Library (SMTL), we can see the list of templates as follows (Fig. 2).
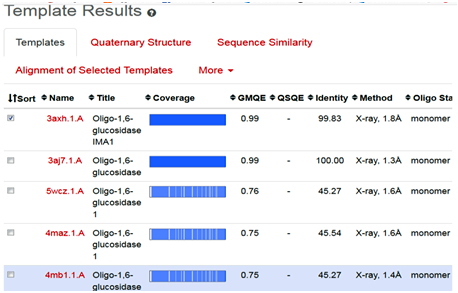
Fig. 2 Depicting the list of template proteins (top-ranked 5) through BLAST and HHblits search program in STML.
When the quick template search is complete, by using combine sequence coverage and sequence similarities, finally a set of maximum 50 top-ranked templates are selected from the complete list of available templates according to a simple score. This furthermore sorted and analyzed according to the expected quality of the resulting models, as estimated by GMQE and QMEAN values. Ranking of templates, on the basis of % identity, GMQE value, X-ray good resolved structures, and the presence of ligand, all are the influencing elements and considerable factors.
In the present study, the topmost, choice in the template list was first one with SMTL I.d (3axh.1A). The other proteins in the list are not containing the ligand glucose; secondly, in the ascending order, they are losing the identity in % similarity and also have increased resolutions. Therefore, template (3axh.1A), is the most suitable, having, excellent % identity (99.83 %), (it’s a rule of thumb, a percent sequence identity greater than 50% is relatively considered for a good modeling project), in our case, most reliable GMQE, was obtained as (0.99) , (as more than 0.7 and near to 1 are more reliable values), the structure was good resolved by X-ray co-crystallography technique with resolution of 1.8 Å , the receptor, is also bound with ligand, which is necessary to trace the binding pocket of enzyme. Therefore, these all are considerable factors, to choose the most suitable template, therefore, we selected template 3ax.1A for homology modeling.
Target & Template Proteins Sequence Alignment
In this step by clicking the arrow in the right, we get the alignment of the target sequence protein (3A4A) and selected template (3axh.1A) protein (Fig. 3).

Fig. 3 Showing the alignment of the target protein (3A4A) with template protein (3axh.1A).
As the above figure is showing excellent alignment between the target and template sequence, with excellent similarity, this we can also observe through sequence similarity in which the target receptor (depicted with a red color circle) is evolutionary so close and identical with the selected template 3.axh.1A. (depicted with a blue color circle). We can also observe the target, template alignments through 3 D models (Fig. 4).
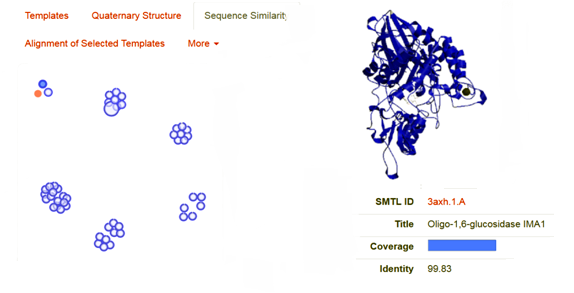
Fig. 4 Showing the sequence similarity b/w target receptor 3A4A (red circle) with selected template 3.axh.1A (blue circle).
Model Building
After the successful alignment model was build up. In SWISS-MODEL WORKSPACE, the quaternary structure annotation of the selected template is used for model building through the sequence of the target in its oligomeric form. This method is established on a machine learning algorithm and Support Vector Machines (SVM), which ultimately combines interface conservation, structural clustering, and other template features to estimate the quaternary structure quality (QSQE), which is ranging from 0 to 1, reflecting the expected accuracy of the interchain contacts required for a model building according to appropriate alignment of template. The value greater than 0.7 of QSQE is considered as better and reliable for quaternary structure prediction in the modeling process. This complements the GMQE score which evaluates the tertiary structure quality accuracy of the model. However, QMEAN is a composite estimator deals with various geometrical properties and provides both local (i.e. per residue) as well as global (i.e. for the entire structure) absolute quality estimation on the basis of a single model, [9-10] (Fig. 5a, 5b, and 5c).

Fig. 5a Showing the 3D structure of developed model visualizing through UCSF Chimera software [11].
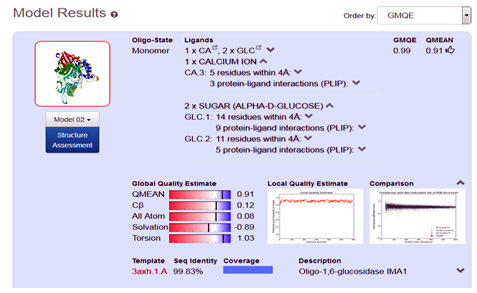
Fig. 5b Showing the Model excellent sequence identity of (99.83%), with GMQE (0.99) and QMEAN (0.91) values respectively.

Fig. 5c Showing the Model and template excellent sequence alignment.
Conclusion
In the present course of study, we develop a 3D structure of the protein α-Glucosidase, by using in-silico based homology modeling technique with the help of most suitable available template in Swiss Modeler Template Library (SMTL). The accuracy of model quality estimation was determined through GMQE and QMEAN value, (0.99) and (0.91) respectively, which shows excellent reliable results. The developed model can be significantly used in drug designing, and drug discovery related projects.
Acknowledgment
The author’s acknowledging to the Swiss Modeler server for its access to accomplish the project successfully.
References
- Biasini M., Schmidt T., Bienert S., Mariani V., Studer G., Haas J., Johner N., Schenk A.D., Philippsen A. and Schwede T. “Open Structure: an integrated software framework for computational structural biology.” Acta Cryst (2013).
- C. Peitsch ProMod and Swiss-Model: Internet-based tools for automated comparative protein Modeling Glaxo Institute for Molecular Biology, I4 chemin des Aulx, I 228 Plan-les-Ouates/Geneva, Switzerland.
- Salentin, S. Schreiber, V.J. Haupt, M.F. Adasme, M. Schroeder PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Research 43 (2015) (Web Server issue):W443-W447. doi:10.1093/nar/gkv315.
- Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F.T., de Beer, T.A.P., Rempfer, C., Bordoli, L., Lepore, R., Schwede, T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46(W1), W296-W303 (2018).
- Berman, H., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig I., Shindyalov I., Bourne P. E. “The Protein Data Bank”. Nucleic Acids Res, 28, 235-242 (2000).
- Berman, H., Henrick, K., Nakamura, H. and Markley, J.L. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res, 35, (2007) D301-303.
- Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K. and Madden, T.L. BLAST, architecture and applications. BMC Bioinformatics, 10, 421-430 (2009).
- Remmert, M., Biegert, A., Hauser, A., and Soding, J. “HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment.”, Nat Methods 9, 173-175(2012).
- Bertoni, M., Kiefer, F., Biasini, M., Bordoli, L., Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Scientific Reports 7 (2017).
- Benkert, P., Biasini, M., Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343-350 (2011).
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis., Comput Chem. Oct;25(13):1605-12 (2004).