
T-Coffee is a multiple sequence alignment tool which stands for Tree-based Consistency Objective Function for alignment Evaluation. It is a simultaneous alignment which combines the best properties of local and global alignment and for this it also uses the Smith-Waterman algorithm. T-Coffee is an advancement over other multiple alignment tools such as ClustalW, MUSCLE (discussed about in earlier article), etc.
Its main features include, first, it provides the multiple alignments using various data sources which is the library of pairwise alignments(global + local). Second main feature is the optimization method which provides the multiple alignment that best fits in the input library.

Fig.1 Layout of the T-Coffee strategy; the main steps required to compute a multiple sequence alignment using the T-Coffee method. Square blocks designate procedures while rounded blocks indicate data structures.
How T-Coffee works?
- Generate Primary library of alignments:
It consists of a set of pairwise alignments of all of the sequences to be aligned (here the alignment source is local). It may also include two or more different alignments of the same pair of sequences. Then the global alignment is done using ClustalW . - Derive primary library weights:
The most reliable residue pair is obtained in this step using a weighted scheme. In this, a weight is assigned to each pair of aligned residues in the library. Here, sequence identity is the criteria to measure accuracy with more than 30 % identity. For each set of sequences, two libraries are constructed along with their weights, one using ClustaW and other using Lalign (program of FASTA package). - Combine Libraries:
In this step, all the duplicated pairs are merged into a single entry that has a weight equal to the sum of two weights, or a new entry is created for the pair being considered. - Extend library:
A triplet approach involving intermediate-sequence method is used. For example, we have 4 sequences, A,B,C & D, it aligns A-B and with C and D as well and checks for the alignment. - Progressive alignment strategy:
In this alignment strategy, a distance matrix is constructed using pairwise alignments between all the sequences, with the help of which a guide tree is constructed using Neighbor Joining (NJ) method (a method that first aligns the two closest sequences), the obtained pair of sequences are checked for gaps,again the next closest two sequences. This continue until all the sequences have been aligned.
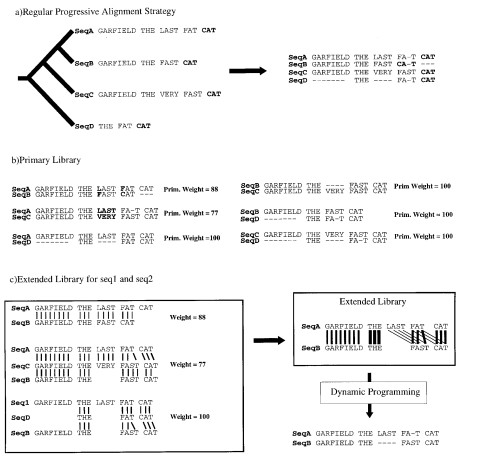
 Fig.2 The library extension. (a) Progressive alignment. Four sequences have been designed. The tree indicates
Fig.2 The library extension. (a) Progressive alignment. Four sequences have been designed. The tree indicates
the order in which the sequences are aligned when using a progressive method such as ClustalW. The resulting alignment is shown, with the word CAT misaligned. (b) Primary library. Each pair of sequences is aligned using ClustalW. In these alignments, each pair of aligned residues is associated with a weight equal to the average identity among matched residues within the complete alignment (mismatches are indicated in bold type). (c) Library extension for a pair of sequences. The three possible alignments of sequence A and B are shown (A and B, A and B through C, A and B through D). These alignments are combined, as explained in the text, to produce the position-speci®c library. This library is resolved by dynamic programming to give the correct alignment. The thickness of the lines indicates the strength of the weight.
Note:
An exhaustive list of references for this article is available with the author and is available on personal request, for more details write to muniba@bioinformaticsreview.com.







