Latest MD Simulation News
MD Simulation using GROMACS: Things to remember
Molecular dynamics (MD) simulation is considered amongst the important methods in bioinformatics.…

Video Tutorial: MD Simulation using GROMACS
This is a video tutorial of our existing article "Molecular Dynamics (MD)…

Tutorial: MD Simulation of a Protein-Ligand Complex using GROMACS
Previously, we have provided a tutorial on molecular dynamics (MD) simulation of…
Installing CHARMM on Ubuntu
CHARMM (Chemistry at Harvard Molecular Mechanics) is a molecular simulation program that…

Tutorial: A quick MD simulation using NAMD and VMD
Molecular dynamics (MD) simulation has become an important methodology in research covering…
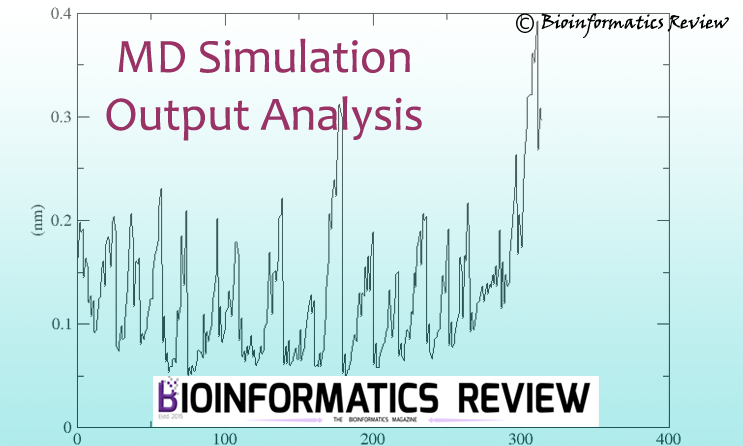
Tutorial: MD simulation output analysis of protein using GROMACS
Molecular dynamics (MD) simulation is an important step in studying the dynamics…