
Installing and executing ProtTest3 on Ubuntu
Prottest3 is a software which is used to select a best-fit amino…

Autodock VinaXB for molecular docking of protein complexes containing halogen bonding interactions
Molecular docking is the most widely used technique which is used to…

How to blast against a particular set of local sequences (local database)?
BLAST is a local alignment tool widely used as a preliminary step…

How to cluster peptide/protein sequences using cd-hit software?
Cd-hit is one of the most widely used programs to cluster biological…

Most widely used tools to analyze multiple sequence alignments
Multiple sequence alignments (MSAs) are quite valuable in terms of studying new…
Installing MODELLER on Linux/Ubuntu
MODELLER is a software package that is used to predict protein three-dimensional…

Video tutorial: How to install Autodock Vina on Ubuntu?
This is a video tutorial of Autodock Vina installation on Ubuntu, based on…

How to perform blind docking using AutoDock Vina?
Blind docking is done when the catalytic/binding residues are unknown in a…
How to calculate dN, dS, and dN/dS ratio on a set of genes using MEGA?
If you want to get a quick idea about the non-synonymous vs…

Installing Roary and Prokka on Ubuntu
In the last article on Bioinformatics Review, the utilization of Roary and…
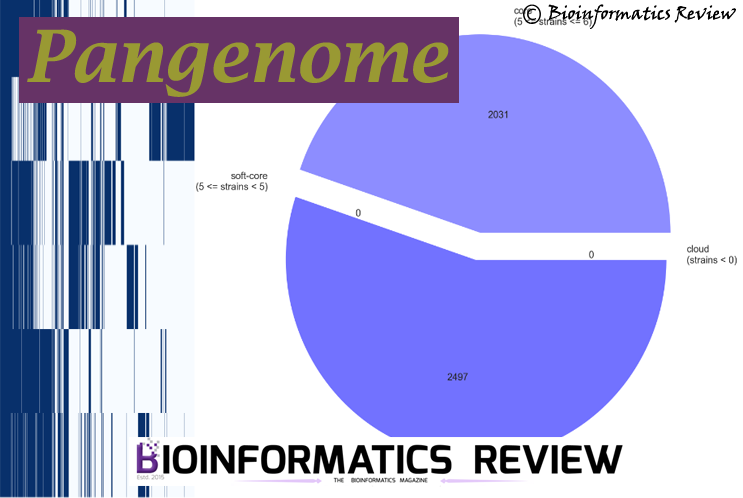
How to create a pangenome of isolated genome sequences using Roary and Prokka?
Roary is a pangenome genome pipeline, which calculates pangenome of a set…
How to do molecular orbital analysis to find d-orbitals involved in bonding in an organometallic compound?
Structure modeling of chemical compounds finds essential application in the field of…

Bioinformatics and stem cell research- A mini review
Stem cells are cells that can be differentiated into other types and…

Web-based tools for protein-peptide docking
Protein-protein interactions are considered necessary in interactome analysis as they play an…
Video Tutorial: How to perform docking using Autodock Vina
This is a video added to our existing tutorial

Most widely used tools for phylogenetic tree customization
Most of the times, it is a very tedious job to convert…

A new high-level Python interface for MD simulation using GROMACS
The roots of the molecular simulation application can be traced back to…

Simulated sequence alignment software: An alternative to MSA benchmarks
In our previous article, we discussed different multiple sequence alignment (MSA) benchmarks…

How to perform protein structure modeling using I-Tasser stand-alone tool?
I-Tasser stands for the iterative threading assembly refinement is a well-known tool…
Intrinsically disordered proteins’ predictors and databases: An overview
Intrinsically unstructured proteins (IUPs) are the natively unfolded proteins which must be…