
An introduction to the predictors of pathogenic point mutations
Single nucleotide variation is a change in a single nucleotide in a…

How to install AutoDock Vina on Ubuntu?
AutoDock Vina is one of the most popular software in Bioinformatics, known…

Bioinformatics Challenges and Advances in RNA interference
RNA interference is a post-transcriptional gene regulatory mechanism to down-regulate…

Recent advances in in-silico approaches for enzyme engineering
Enzymes are natural biocatalysts and an attractive alternative to chemicals providing improved…

Lipidomics: An overview
Lipids are the essential metabolites in human body performing different functions such…

A short introduction to protein structures modification and ModFinder
A lot of protein structures are determined on a large scale and…

Alignment-free approaches for Sequence Analysis
Multiple Sequence Alignment (MSA) is a fundamental aspect of Bioinformatics in order…

How to perform docking in a specific binding site using AutoDock Vina?
AutoDock Vina is a bioinformatics tool that is used to perform in-silico…
Methods to detect the effects of alternative splicing and transcription on proteins
Alternative splicing and the transcription are the most familiar processes amongst the…

PcircRNA_finder: Tool to predict circular RNA in plants
The non-coding circular RNAs (circRNA) play important role in controlling cellular processes.…
Prediction of Protein-Protein Binding Affinity through their Amino Acid Sequence
Protein-protein interactions (PPIs) have become necessary in order to study many biological…
miRNAs and their Target Prediction Tools: An Overview
miRNAs are the small endogenous non-coding RNAs having a length less than…
DrugQuest: Tool for Drug-associated Queries
In this rising era of personalized medicine, drug discovery, data refining, chemical…
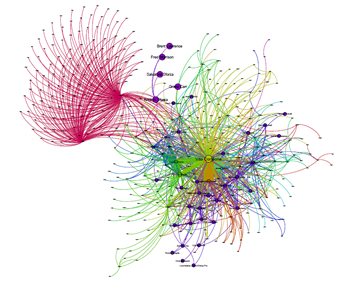
Cytoscape.js : A graph library for network visualization and analysis
Network visualization has become a strong need for studying the molecular interactions…
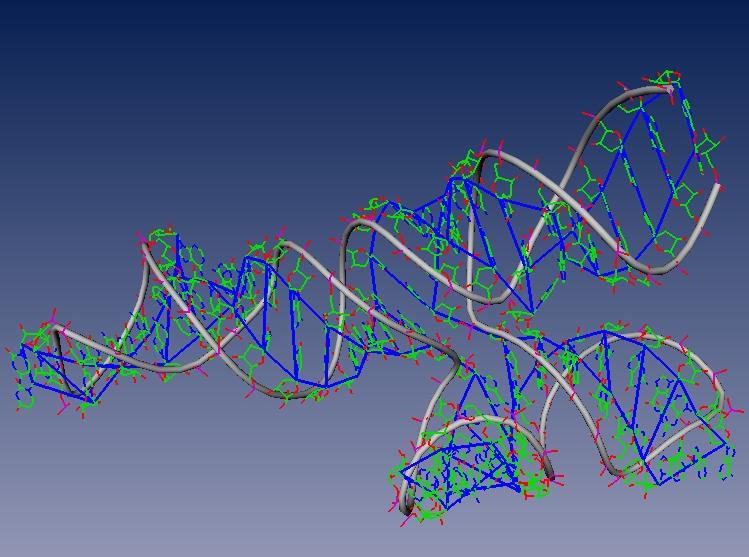
Foldalign: a tool for secondary structure alignment of RNA
Secondary structure formation and conformational changes play a key role in understanding…
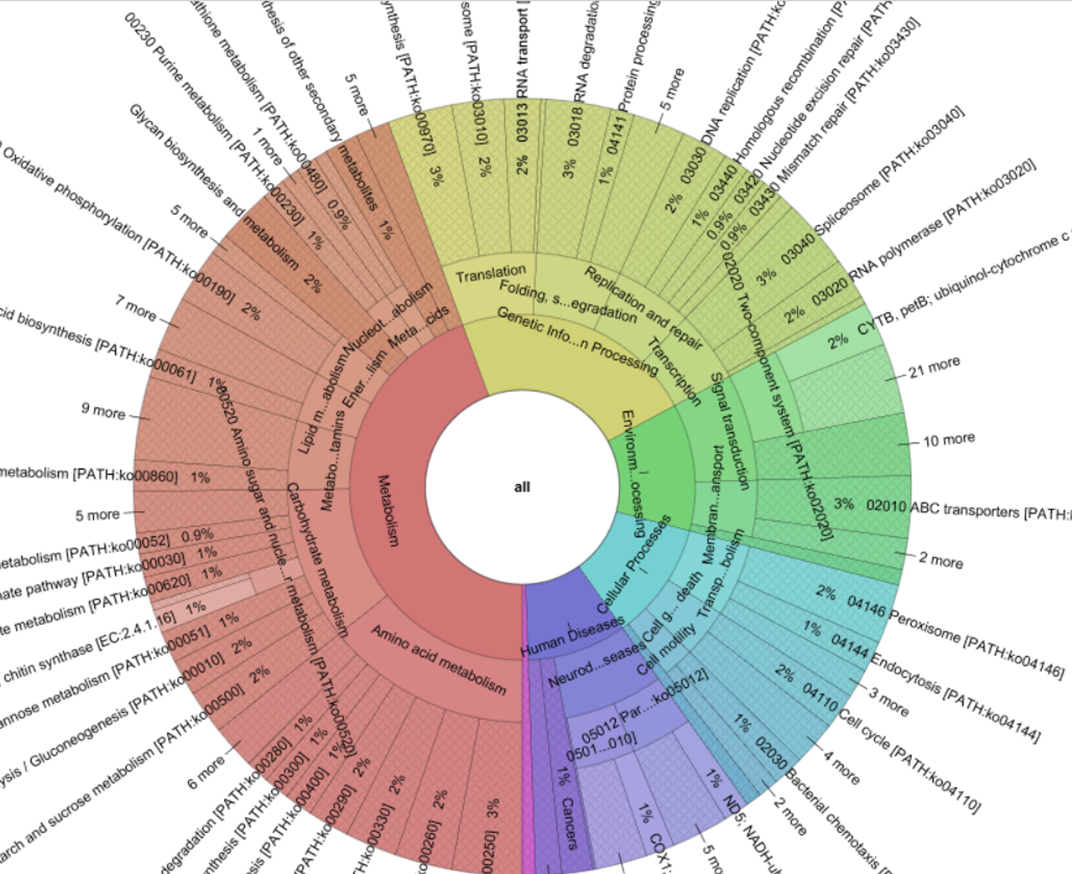
Predictive metagenomics profiling: why, what and how ?
What is predictive metagenomics profiling? Recently, predictive metagenomics profiling (PMP) has been…

A practical guide to selection analyses of coding sequence datasets and its intricacies
This is the second article under the popular series “Do you HYPHY…”,…
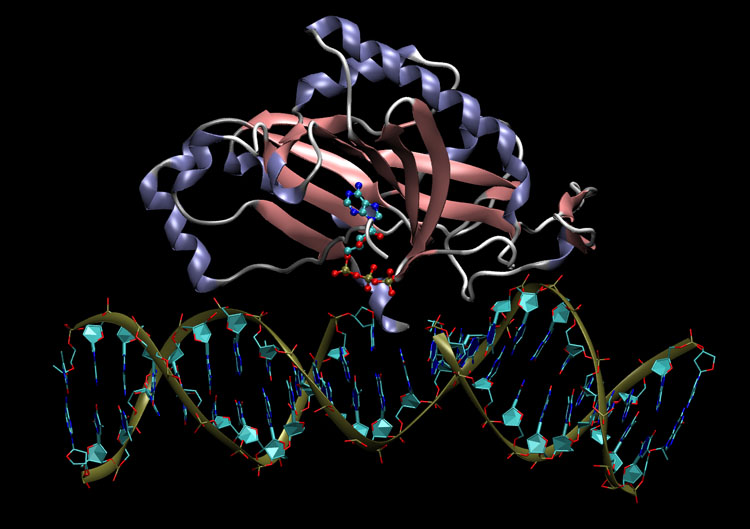
WebFEATURE : Tool to identify and visualize Functional Sites in Macromolecules
The identification and assignment of functions of unknown macro molecules has been…

MUSCLE v/s T-COFFEE : An overview and different aspects
As I have discussed in my earlier articles about the multiple sequence…

T-Coffee : A tool that combines both local and global alignments
T-Coffee is a multiple sequence alignment tool which stands for Tree-based Consistency…