Latest Molecular dynamics News

How to generate PSF file for MD simulation using NAMD & VMD?
Protein structure file (PSF) contains topology information and is required for molecular…

How to create an index file in GROMACS for MD simulation?
MD simulation is a tricky technique if you don't understand what you…

Tutorial: MD Simulation of a Protein-Ligand Complex using GROMACS
Previously, we have provided a tutorial on molecular dynamics (MD) simulation of…
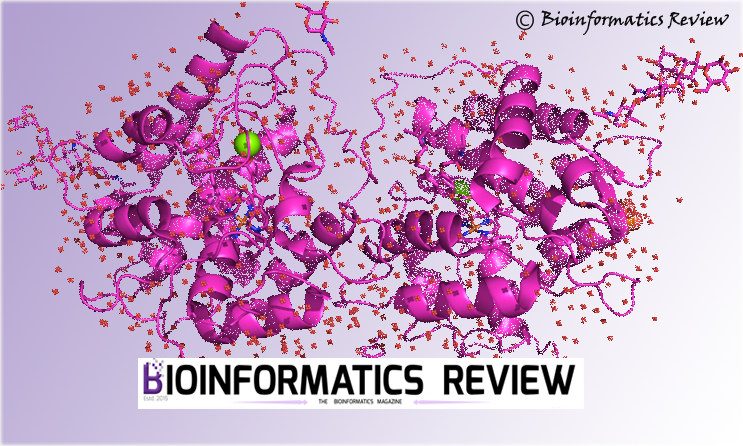
Installing CHARMM on Ubuntu
CHARMM (Chemistry at Harvard Molecular Mechanics) is a molecular simulation program that…

Video tutorial: Installing GROMACS on Ubuntu
This is a video tutorial of our existing article on GROMACS (Abraham,…

Tutorial: Molecular dynamics (MD) simulation using Gromacs
Gromacs is one of the most widely used software for molecular dynamics…

A new high-level Python interface for MD simulation using GROMACS
The roots of the molecular simulation application can be traced back to…

Installing Gromacs on Ubuntu for MD Simulation
In bioinformatics, GROMACS is one of the most popular Molecular Dynamics simulation…