
How to concatenate FASTA sequences using Perl?
Here is a simple Perl script to concatenate multiline FASTA sequences into…

How to search motif pattern in FASTA sequences using Perl hash?
Here is a simple Perl script to search for motif patterns in…

A perl script to convert multiline FASTA sequences into a single line
There are different software or tools which require different kinds of input,…

How to read fasta sequences from a file using PHP?
Here is a simple function in PHP to read fasta sequences from…

How to extract fasta sequences from a multi-fasta file based on matching headers in a separate file?
This is a simple Perl script to extract FASTA sequences from a…

How to read fasta sequences as hash using perl?
This is a simple Perl script to read a multifasta file as a…

BETSY: A new backward-chaining expert system for automated development of pipelines in Bioinformatics
Bioinformatics analyses have become long and difficult as it involves a large…

Linux ‘sed’ command in Perl programming
When it comes to handling large data files to process, it becomes…

How to execute Unix/shell commands in a Perl script?
BioPerl is a collection of Perl modules that are used to write…

What is Numerical Taxonomy? How is it useful?
Classification of biological species is one of the important concern while studying…

Genetic Algorithm: Explanation and Perl Code
When it comes to bioinformatics algorithms, Genetic algorithms top the list of…
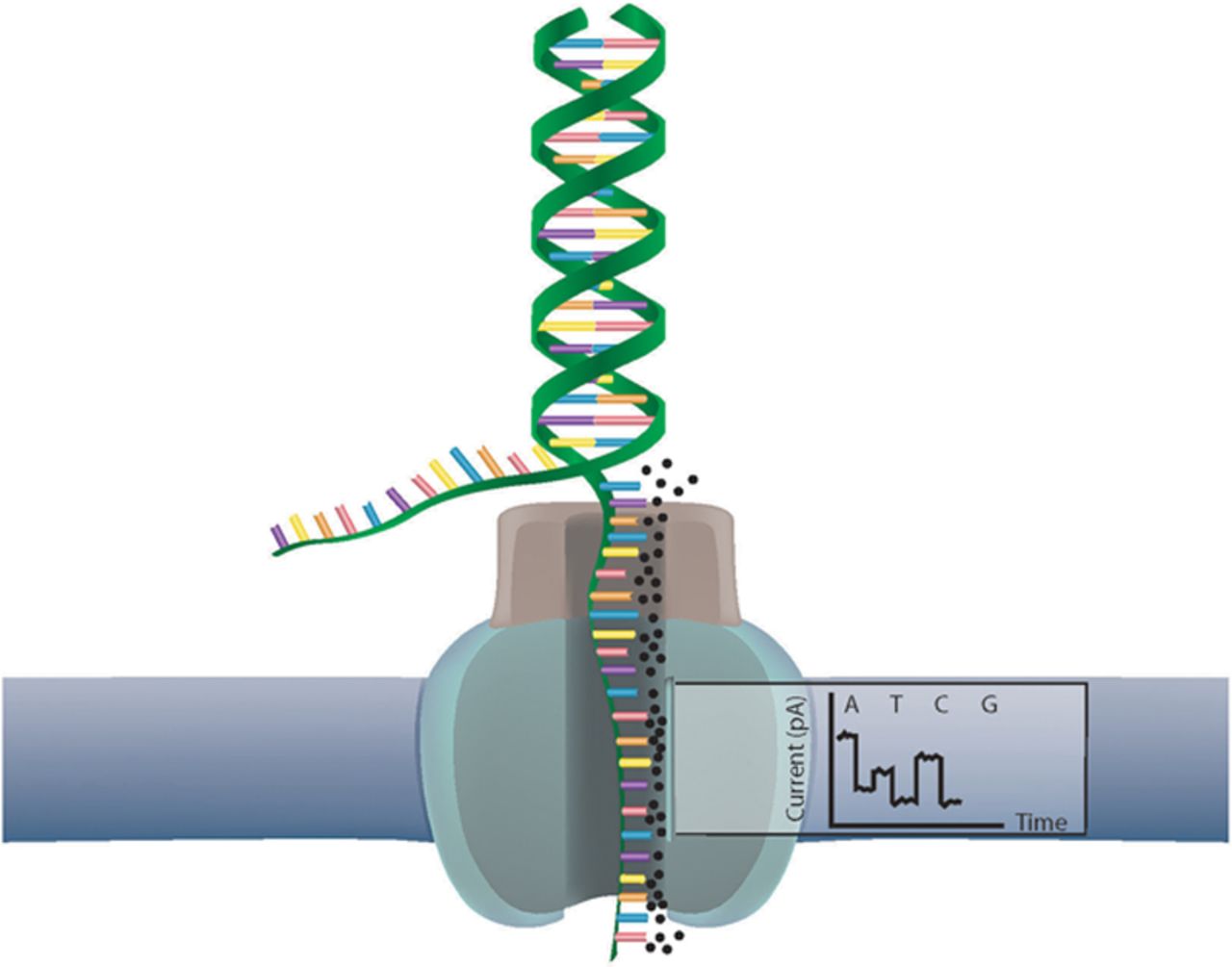
HTSeq : A Python framework to analyze high throughput sequencing data
High throughput sequencing is most widely used as it saves a lot…

TIN: R package to analyze Transcriptome Instability
Alternative Splicing plays a very essential role in proper functioning of eukaryotic…

Perl one-liners for bioinformaticians
Perl one-liners are extremely short Perl scripts written in the form of…