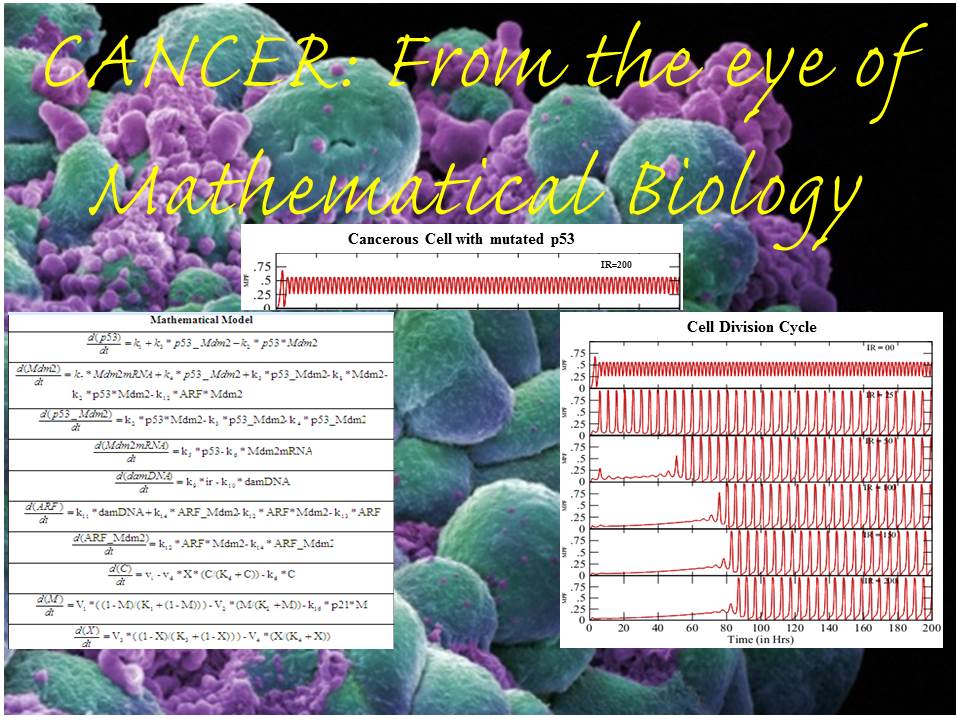
Cancer has become a very common disease now a days, but the main reason of causing this is unknown up till now. Various reasons have been given and recent research says that improper sleeping patterns may also lead to cancer. Like cause of cancer is difficult to predict, similarly, its progression and prognosis is also very difficult. Despite of many advances in cancer treatment, early detection is still very difficult. While there have been many early cancer screening techniques, but are not realistic because of the lack of cost-effectiveness or requirement of invasive procedures. Genomic screening techniques are a promising approach in this area.
Gene expression signatures are commonly used to create cancer prognosis and diagnosis methods. Gene expression signature is a group of genes in a cell whose combined expression pattern is uniquely characteristic of a biological phenotype or a medical condition.
But only few of them were successfully able to utilize in clinics and many of them failed to perform. Since these signatures attempt to model the highly variable and unstable genomic behavior of cancer, they are unable to predict cancer. The degree of deviation in gene expression from the normal tissue, i.e., the hyper-variability across cancer types can be used as a measurement of risk of relapse or death. This gives rise to the concept of Gene expression anti-profiles. Anti- profiles are used to develop cancer genomic signatures that specifically takes advantage of gene expression heterogeneity. They explicitly model increased gene expression variability in cancer to define robust and reproducible gene expression signatures capable of accurately distinguish tumor samples from healthy controls.
Differentially variable genes= anti-profile genes
After employing many experiments regarding cancer anti-profiles, the results indicated that the anti-profile approach can be used as a more robust and stable indicator of tumor malignancy than traditional classification approaches.
The researchers’ hypothesis is that the degree of hyper-variability (w.r.t normal samples) is directly proportional to the tumor progression, i.e., degree of hyper- variability as measured with respect to the normal samples would increase with tumor progression. Corrada Bravo et al found out a way to derive a colon-cancer anti-profile for screening colon tumors by measuring deviation from normal colon samples. To create an anti-profile, they used a set of normal samples and tumor samples, probe- sets are then ranked by the quantity σj,tumor/ σ j,normal(where σj,tumor and σj,normal are the standard deviations among the tumor samples and normal samples, respectively, for probeset j) in descending order, and a certain number of probesets (typically 100) with the highest value are selected. Then they calculated the normal regions of each probe set and then the number of probe sets for which the expression lies outside the normal region was calculated to get an anti-profile score of the sample.
To test their hypothesis, they obtained two publicly available microarray datasets with normal, adenoma, and cancer colon samples. By studying these datasets, they plotted the distribution of variance of cancer/adenoma samples to variance of normal samples ratio (in log2 scale) for these probe sets on the other dataset (Fig. 1A and B).

Fig.1 Among probes that exhibit higher variability among cancers than among normals, the degree of hypervariability observed is related to the level of progression. (a) Distribution of variance ratio statistic {log2 σ σ2tumor ÷ σ2normal} for colon dataset (Gyorffy et al; GSE4183) from anti-profile computed using another colon dataset (Skrzypczak et al; GSE20916). (B) Distribution of variance ratio statistic for Skrzypczak et al colon dataset from anti-profile computed using Gyorffy et al colon dataset. (C) Distribution of variance ratio statistic for adrenocortical data (Giordano et al; GSE10927) for universal anti-profile probe sets.
Both adenoma and cancer samples show higher variability than normals (region to the right of x = 0), while cancer samples show higher hypervariability than adenomas. This suggests that hypervariability is a stable marker between experimental datasets and that specific selection of hypervariable genes across cancer types and the anti-profile method can be extended to model tumor progression. These studies showed that Gene expression anti-profiles capture tumor progression.
DNA methylation is one of the primary epigenetic mechanisms for gene regulation, and is believed to play a particularly important role in cancer. High levels of methylation in promoter regions are usually associated with low transcription. Cancer has loss of sharply methylation levels which is associated with increased hypervariability in gene expression across multiple tumor types. They applied the anti-profile scoring method to DNA methylation data from thyroid and colon samples, where for each tissue type, normal, adenoma and cancer samples were available. Figure 2 shows the distribution of adenoma and carcinoma samples against normal samples on a principal component plot, showing the presence of the hypervariability pattern in methylation data: the normal samples cluster tightly, while the adenomas show some dispersion and the carcinomas show even greater dispersion. Since these behaviors are present for both colon and thyroid data, it again reinforces their notion that the anti-profile approach has wide application for classification in cancer.
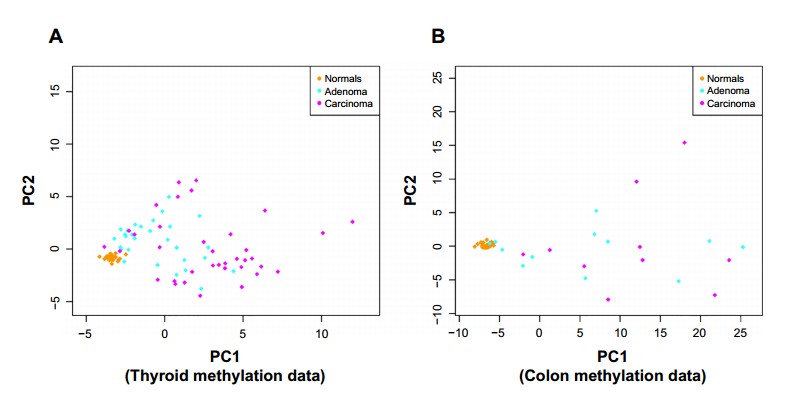
figure 2. Anti-profiles applied to methylation data: first two principal components of (A) thyroid methylation data and (B) colon methylation data.
Conclusion:
The anti-profile approach is more suitable for cancer prognosis. It can robustly predict the tumor progression and prognosis based on the variability in the gene expressions. The results presented above also confirms that gene expression signatures based on hyper-variability can be highly valuable.
References:
Wikum Dinalankara and Héctor Corrada Bravo Center for Bioinformatics and Computational Biology, Department of Computer Science and UMIACS, University of Maryland, College Park, MD, USA