
This is a video tutorial of our existing article “Molecular Dynamics (MD) Simulation using GROMACS“.

Previously, we have provided a tutorial on molecular dynamics (MD) simulation of a protein using GROMACS [1] and its result analysis [2]. In this article, we...
CHARMM (Chemistry at Harvard Molecular Mechanics) is a molecular simulation program that can be used for simulation of macromolecules, complexes, and many-particle systems [1]. In this...

Molecular dynamics (MD) simulation has become an important methodology in research covering systems consisting of millions of atoms. We have provided several articles on MD simulation...
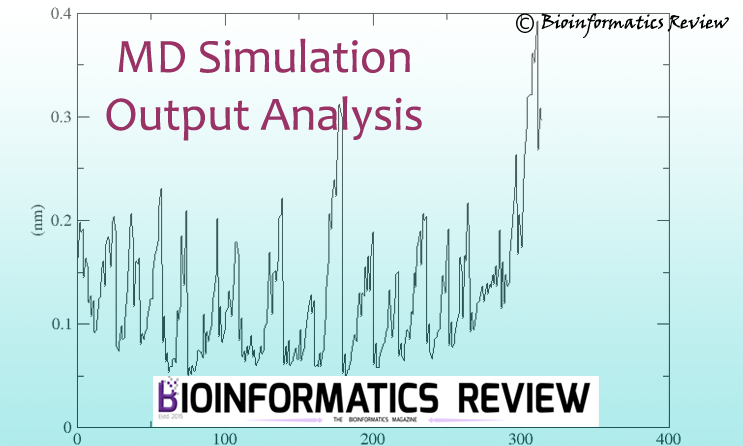
Molecular dynamics (MD) simulation is an important step in studying the dynamics of macromolecules. In one of the previous articles, MD simulation of chain A of...
Latest Comments