Homology Modeling
Methodology for Homology Modeling of a Simple Protein
Previously, we have explained the initial methods involved in the structure prediction of a protein. In that article, we discussed the three basic methods involved in protein structure prediction: Homology modeling, ab-initio, and threading. In this article, we will explain the methodology involved in performing the homology modeling of a simple protein.
Homology modeling uses a template structure to predict a new structure for a query protein. We go for homology modeling when enough similarity (~ 30-32 %) for the query protein can be found in the available databases. The most widely used and accurate prediction server is the Swiss Model [1]. A complete flowchart is shown below in Figure 1.
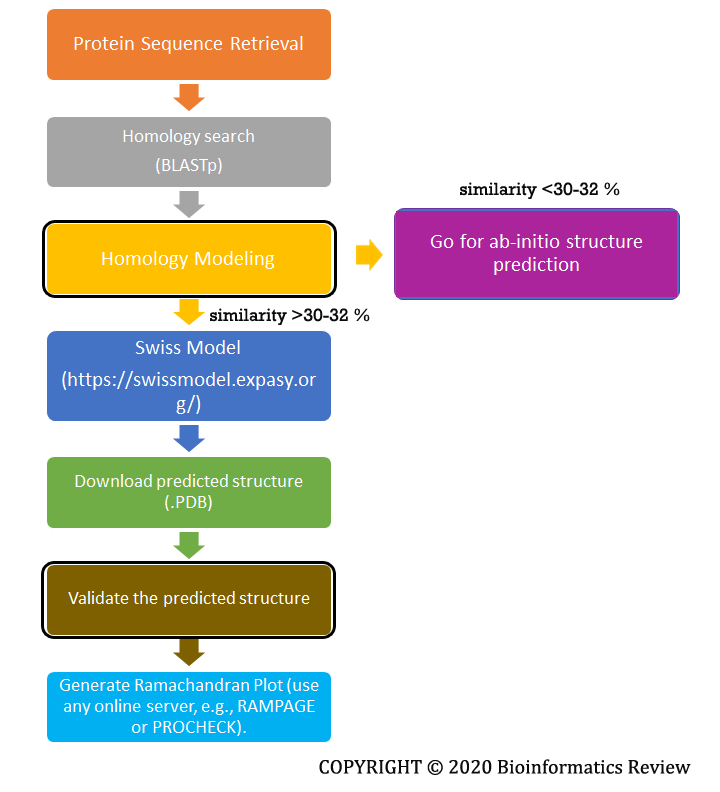
Figure 1 Basic steps involved in homology modeling.
These are the basic steps followed during the homology modeling of a simple protein. There are several other software/tools that are used for homology modeling but the Swiss Model provides the most accurate results. Ab-initio prediction will be explained in upcoming articles.
References
- Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., … & Lepore, R. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic acids research, 46(W1), W296-W303.












SALI MODELLER [1] is one of the most widely used command-line bioinformatics software for protein structure prediction based on homology modeling. The installation of MODELLER on Ubuntu has already been explained in an article published previously. This article will explain how to perform basic modeling of a protein sequence having a high percent identity with the template. (more…)

You must be logged in to post a comment Login